(2023年9月18日更新) [ 日本語 | English ]
HOME > 講義・実習・演習一覧 / 研究概要 > 小辞典 > 熱力学
|
物質の分子構造とは無関係な形で、物質の熱に関する性質をまとめる
エネルギー原理 energy principle: 物理現象をエネルギーにより解析 Def. エネルギー伝達 energy transfer: 断面通過するエネルギー流 flux種々現象に適用可能 - その時の(状態方程式と)ΔWの表現を検討必要 Def. 量変化記号: Δ: 有限変化 > δ: 微小(量)変化 > d; 瞬間変化Ex. Q: 熱量(比熱) → ΔQ, δQ, dQ 状態量 state quantity or property≡ その時の状態で決まる量 = 示量性状態量 + 示強性状態量⇔ 非状態量 Ex. Q, W (系に外からする仕事) ⇒ 物理量Xが状態量にある系 → 系が状態Aから状態Bに変化したときΔXは状態変化経路に依存しないEx. T, V, P, L, f, U, S, F, G, H, 電気, 磁気(エネルギーに似る: 要基準点) UI → UII (T, P, V) ⇒ ΔU = UII - UI (微小変化ではδU = δQ - δW) 熱力学関数 thermodynamical functions分子質量 molecular mass示量性 extensive property: 系の質量に比例する量 Ex. 熱量 Q, 体積 V, 内部エネルギー U, 磁化 M, 分性率 P 示強性 intensive property: 系の質量に依存しない変数(エネルギー多化)Ex. 温度 T, 圧力 P, 密度 D, H, E, f ⇒ ΔWを表わす変数としては示強変数と示量変数とが対になり現れる
ΔW = -PΔV = fΔl R ≡ 1 K気体温度上昇時に膨張のため気体が外にする仕事 熱量計 calorimeter: 熱量を計測する機器熱力学史 (history of thermodynamics)Boyle 1627-1691, 英: ボイルの法則(1660) ⇒ PV = RT「懐疑的な化学者」(1680): 4元素説、3元素説否定 – 元素は実験的分析により決められる(当時は否定的) Mariotte 1620-1684, 仏: 流体・気体研究1676 ボイル・シャルルの法則(マリオットの法則)提唱 気圧計用い天気予報の基礎作る Becher 1635-1682, 英当時までの英国医学の物質要素概念: 可燃性原質(硫黄)、流動性原質(水銀)、固定性原質(塩) 金属灰化過程 – 既概念を修正し、固体の土性物質の含む成分の変化で説明
1) 固定された土 → 鉄粉と硫黄の混合物 = 「レムリーの火山」と呼ぶ Lémery, Louis 1677-1743, 仏(Nicolas息子): 化学仏国王侍医。親子共に仏科学アカデミー会員 Stahl, Georg Ernst 1660-1734, 独: 化学・物理学・哲学1703 フロギストン説 phlogiston theory
可燃性物質燃焼時に、油性の土(= フロギストン)が空気に出て行き、灰が残る – ギリシア4元素説否定 1754 二酸化炭素(固定空気)発見 → 空気と異なる 炭酸塩が塩基と固定空気の複合物であることから発見 Cavendish, Henry 1731-1810, 英(物理・化学)1766 電気・熱研究、地球質量・比重測定等も行う
金属 + 酸 → 水素(可燃性空気) 1772 窒素(フロギストン化空気)発見 Scheele Carl Wilhelm 1742-1786, 英: 薬化学 pharmaceutical chemistry 1774 塩素発見(脱フロギストン塩酸) 1777 酸素(微細な酸性物質 + フロギストン) – 実際はPriestleyより先に発見 ⇒ 発見気体増 → フロギストン説の不備指摘 Priestley, Joseph 1733-1804, 英: 化学・聖職(フィラデルフィアに米国最初のユニテリアン教会設立) 1774 燃焼: 空気の一部が燃える物質と結合1775 酸素(脱フロギストン空気)
酸素、窒素、二酸化窒素等、各種ガスを発見分類 蒸気機関発明者の1人 → 産業革命 = 石炭(地学進展) 複動回転蒸気機関製作 – Newcomenの機関を元に作成 表. 産業革命期の発明 • 1765 Watt J: 蒸気機関 • 1782- Watt J: 往復動機関 • 1766 Roy, P Le 仏: クロノメーター(航海用精密時計) • 1768 Arkwright R 英: 水力紡績機械 • 1787 Cartwright E 英: 力織機 • 1802 Trevithick R 英: 蒸気車 • 1807 Fulton R 米: 汽船 • 1814 Stephenson G 英: 蒸気機関車 (1825 Stockton-Darlington Railway) |
Ex. Wattはグラスゴー大学Black J (1728-1799)協力で研究進む Lavoisier 1743-17941789 Traité Elémentaire de Chimie (化学要綱) – 初の近代的化学教科書 C + O2 = CO2 (実験的証明) → ラボラジエの質量保存則(熱素caloricを反応概念とし元素中に加える) 空気は窒素と酸素からなる(空気単一成分説否定) → 酸素が燃焼に関与 Graf von Rumford 1753-1814, 米 → 英・独砲身に穴を開ける際の発熱を研究 – 熱素説否定 Proust, Joseph Louis 1754-1826 定比例の法則law of definite proportions (例外: 同位元素・欠陥格子) ⇒ Dalton, John 1766-1844, 英 1808 A new system of chemical philosophy (化学の新体系) ダルトンの原子説 Avogadro A 1776-1856, 伊 分子説 = 物質は同種・異種原子が結合した分子 → アボガドロの法則 Gay-Lussac, Joseph Louis 1778-1850 ゲイリュサックの法則(= シャルルの法則) 1808 気体反応の法則 law of gaseous reaction 2種類気体が化合し、生成物が気体である時、2種類の気体及び生成物の容積は簡単な整数比となる 1810 アルコール発酵化学式Carnot, Nicolas Leonard Sadi 1796-1832, 仏 熱現象(熱機関)効率を原理的に解明 → 熱力学の基礎を築く エネルギー保存則を考慮した"熱素説": 熱エネルギーと力学的エネルギーを同一体系に組み入れず熱は熱素とし保存する(誤り) [カルノーサイクルのように概要は知っていた] Krönig, August 1822-1879: 気体の気体分子運動論 (1856) Clausius, Rudolf Julius Emanuel 1822-1888, 独: 熱力学の祖 カルノー体系を熱力学とし確立 = 力学的Eは熱Eに変換可能なことを示す Gibbs, Josiah Willard 1839-1903, 米: 物理学・化学 熱力学の理論的体系の基礎を作る Clapeyron, Emile 1799-1864, 仏: 熱力学 Kelvin, William Thomson, 1st Baron 1824-1907, 英: 熱学・電磁気学 (絶対温度単位Kは彼の名) von Mayer, Julius Robert (1814-1878, 独) 1842 エネルギー保存則 = 熱 ↔ 仕事 Joule, James Prescott 1818-1889, 英 (エネルギー単位Jは彼の名) 電流発熱作用発見法則化 1843 熱仕事当量実測 - エネルギー保存則 Helmholtz, Hermann LF 1821-1894 1847 「エネルギー保存則」定式化 Hittorf, Johann Wilhelm 1824-1914, 独 電解中イオン移動研究から輪率概念提出、電気化学の基礎築く Arrhenius, Svante August 1859-1927, Sweden 物理化学創始者の1人 (ノーベル化学賞 1903) 電離理論。イオン説確立
|
機関 (engine)≡ 動力用の装置外燃機関 (external combustion engine) ≡ 機関外で燃料を燃焼させる機関 Ex. 蒸気機関 ⇔ 内燃機関 (internal combustion engine) ≡ 機関(シリンダ)ー内で燃焼
燃料噴射 (fuel injection)
Ex. 蒸気機関、ガソリンエンジン、ディーゼルエンジン 燃焼 (combustion)定温度以上に保たないと継続しない = 放射・対流・伝導で散逸する熱より発生燃焼熱が上回る必要
(通常) 燃焼に必要な酸素は大気中から供給 → 可燃物の表面・形状・周囲状況により変化 鉄: スチールウールはライター等で着火可能 → 鉄塊は火を近づけただけでは燃焼しない |
発火 (点火) (ignition) ≡ 物体自身が燃え出すこと 可燃物 (combustible matter) ≡ 通常環境で着火するもの → 燃焼継続する物体 ⇒ 可燃性 flammable 発火点(着火点) (kindling temperature): 物質を空気中で加熱→ 発火し始める最低温度 引火点 (flash point): 炎存在 = 着火最低温度(燃焼継続しなくて良い)
(s.s.) 液体の可燃物が液面から爆発限界の最低値の濃度の蒸気を発生させるのに足りる最低温度 可逆過程(機関) reversible process (engine) ↔ 不可逆過程(機関) irreversible process (engine) 準静的変化 → 熱効率 heat efficiency // Clausiusの原理 → エントロピー 熱 = 仕事: 同等 ⇒ 1 cal = 4.1868 J (定義される), 1 W = 1 J/s Def. 熱の仕事等量(当量) machanical equivalent of heat: 熱と仕事との比⇒ 4.19 J/cal コジェネレーション cogeneration: 1エネルギーから複数エネルギー(電気・熱等)を取出すシステム |
気体の持つエネルギーDef. 運動エネルギー: 気体が動いていることによる, Ek = (1/2)mv2Def. 位置エネルギー: 気体高い位置にあることによる, Ev = mgz Def. 内部エネルギー: 気体の空気分子の運動による, U = CvT 系 [平衡系] → 熱の出入り ≡ エネルギー変化 Law 熱力学の第0法則 zeroth law of thermodynamics≡ 長時間後に孤立系は熱平衡状態に達する 熱平衡: 温度の異なる物体を接しておく → 長時間後に両者が一様な温度となり変化止まる→ 2つの物体は熱平衡状態 thermal equilibrinum state にある Law 熱交換法則 law of heat exchange: ΣQout + + ΣQin = 0Law 熱力学の第1法則(エネルギー保存則) (数学的表現)ΔU (Uという状態量を表す) = ΔQ + ΔWQ: 系外部からの熱エネルギー W: (行った)仕事エネルギー Q - W = U2 - U1 (U1: 熱を加える前のU. U2: 熱を加えた後のU)⇒ 加えた熱の一部を仕事Wに、残りを内部エネルギーU変化に使う ΔQ = ΔW + ΔU (第1法則式)⇒ 系外部からの熱 = 気体のした仕事 + 内部エネルギー変化 Ex. 気体の体積変化 + 内部エネルギー変化 ⇒ 理想気体の内部エネルギーは温度のみの関数 = 体積に無関係 巨視的に見た熱力学第一法則: 巨視的に観測できる量(↓)のみで記述可能
P: 圧力, T: 温度, V: 容積, Q: 熱量, W: 仕事 系 systemの全エネルギーtotal energy ≡ 内部エネルギーinternal energy, l = U = Ef + Eb 外界 external worldとの交換 熱 物質 仕事 孤立系 isolated system: × × × U = constant ⇒ ΔU = 0 外界から遮断 断熱系 adiabatic system × × O ΔU = -W閉鎖系 closed system O × O ΔU = Q – W 開放系 open system: O O O 外界と全て交換 記号の定義: W, 仕事. Q, 熱開放
気体の一般法則 general gas law実在気体 → 理想化 → 理想気体(実在気体でも減圧 reduced pressure → 低温度領域では理想気体に近づく)Def. 標準状態 (normal or standard state): 標準圧力温度での気体の状態
≡ NTP or STP (normal temperature and pressure) ※ 熱力学関数では25°Cを標準状態とする 標準状態での気体: 1 l = 2.69·1022個 vs 1 mol = 6.02·1023個(6.02·1023)/(2.69·1022) = 22.4 l ⇒ 22.4 lの標準状態気体は1 mol |
Q. 空気: N2:O2 = 4:1 (v/v)混合物 ⇒ 空気の平均分子量(みかけの分子量) A. 空気22.4 l = 1 mol ⇒ N2質量 = 28 g × 4/5, O2質量 = 32 g × 1/5 ∴ 空気の平均分子量 = 22.4 + 6.4 = 28.8 g
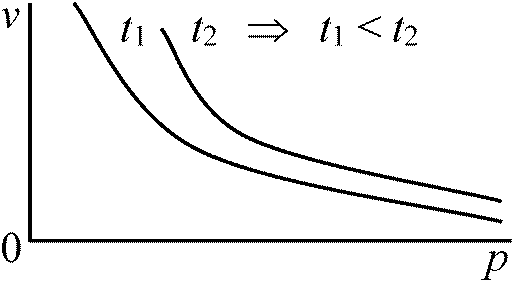
Law. ボイルの法則 Boyle's law (1662)一定質量気体: 体積 ∝-1 圧力t = constant, isotherm, p-v relationship: V ∝ 1/p ⇒ V = k·1/p, k: 比例定数 Law. シャルルの法則 Charles's law= ゲイリュサックの法則 Gay-Lussac's law気体の熱膨脹率は一定
p = constant: m (質量) = constant, 0°C → v0, t°C → vt Law. ボイル・シャルルの法則 Boyle-Charl's law一定質量の気体の体積: ∝ 絶対温度, ∝-1圧力pv = nRT, p: air pressure (N/m2), v: volume (m3)
T: 絶対温度(K)
R = pv/T = (1 × 22.414)/273.16 = 0.08205 latmK-1mol-1 ⇒ Def. 理想気体 ideal or perfect gas: ボイル-シャルルの法則成立する気体
成立条件: 圧力 ∝ 温度・密度 (実在しない) 水上捕集: 大気圧(P) = 気体の真の圧力(Pa) + その温度の水蒸気圧(Pw) 分子量測定法
≡ 混合気体の全圧Vは成分気体の分圧の和に等しい 互いに影響を及ぼさない複数気体, Vi ⇒ V = V1 + V2 + … = ΣVi (加成性 additivity) → (P1 + P2 + …)V = P(V1 + V2 + …) = (n1 + n2 + …)RT ∴ (P1 + P2 + …)V = PV = (n1 + n2 + …)RT ⇒ P = P1 + P2 + … = ΣPi, ΣPiV = PΣVi = RTΣni ∴ ΣPi = Ppi/p = xi ⇒ pi = pxi (≡ モル分率 mole fraction)
ni/(n1 + n2 + …) = xi |
気体 gasDef. 拡散 diffusion: 境界面を通し一方の気体が他の気体中に混ざって行く現象 - 分子速度に比べ非常に遅い
物質が熱平衡状態に近づく際に起こる濃度分布変化過程 分子速度 ∝ 衝突数、密度densityに比例 → uavg = √(8kT/πm) Law. Grahamの法則(実験的発見)⇒ 同条件(=圧力)の元で気体流出速度 ∝-1 √(その気体密度)
⇒ Gas 1, 2, 流出時間t1, t2, 流出速度E1, E2, 分子量 M1, M2 力学的気体の分子運動論 kinetic theory of gases気体分子の分布: 気体分子運動 ⇒ 仕事をせず移動 vs 仕事をして移動
原子論的atomisticアプローチ ⇒ 壁で気体分子の運動量変化 (= 反作用) (Krönig, Clausius) Eq. ベルヌーイ公式: 立方体の各面の受ける圧力の変化 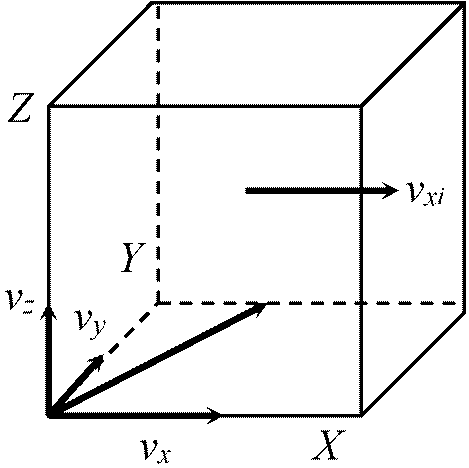 Ex. 立方体1 cm3, n = 1 (cm3)が含む分子数
Ex. 立方体1 cm3, n = 1 (cm3)が含む分子数
v2 = vx2 + vy2 + vz2 … (1) (v 速度, vx, vy, vz 各軸成分) vxi: x軸に沿い運動 → yz面に当る: 運動量(+vxi → –vxi) → 2vxi運動量変化
→ 1 sec: fxi = (vxi/2)·2mvxi = mvxi2 気体分子運動は任意の方向 at random Σi=1nvxi2 = Σi=1nvyi2 = Σi=1nvzi2 Def. 平均2乗速度 mean square velocity, 〈v2〉 = (Σi=1nvi2)/n⇒ 〈v2〉 = 〈vx2〉 + 〈vy2〉 + 〈vz2〉 = 3〈vx〉 PV = 1/3·nm〈v2〉 = 2/3·E,
m: 質量, n: 分子数(分子密度) ∴ E = 2/3·RT ⇒ 各分子が持つ運動エネルギーの和 → 気体1個あたりの運動エネルギー, E1 = 1/2·m〈v2〉 = 3/2·R/NA·TDef. ボルツマン定数 Boltzmann constant, kB = R/NA ≈ 1.38 × 10-23 J/K ∴ E1 = 1/2·m〈v2〉 = 3/2·kB·T ⇒ ボイル-シャルルの法則導出 速度vの分布関数1 cm – n個 … nf(vx)
Aに分子が存在する確率: dNA = nf(vxA)f(vyA)f(vzA)dvxdvydvz 分子の存在を全体的に表わす ⇒ 球対称 ⇒
dn = f(vx)f(vy)f(vz)dvxdvydvz  点Aとすぐ近くのA'との間の分布関数: A'(δvx, δvy, δvz)
点Aとすぐ近くのA'との間の分布関数: A'(δvx, δvy, δvz)f(vxA + δvx)f(vyA + δvy)f(vzA + δvz) - f(vxA)f(vyA)f'(vzA) = f'(vxA)f(vyA)f(vzA)δvx
+ f(vxA)f'(vyA)f(vzA)δvy = φ'(vA)δv AとA'が同一球面上 ⇒ fv = 0f'(vxA)f(vyA)f(vzA)δvx + f(vxA)f'(vyA)f(vzA)δvy + f(vxA)f(vyA)f'(vzA)δvz = 0 ⇒ 一般化(両辺をf(vx)f(vy)f(vz)で割る) f'(vx)/f(vx)·δvx + f'(vy)/f(vy)·δvy + f'(vz)/f(vz)·δvz = 0 … (1) 球対称の点Aに対する速度vA: (vA)2 = (vxA)2 + (vyA)2 + (vzA)2 点A'に対する速度vA': (vA')2 = (vxA' + δvx)2 + (vyA' + δvy)2 + (vzA' + δvz)2 右辺の( )内を展開しδvx2等の項を ≈ 0として無視すると (vA')2 - (vA)2 = 2vxAδvx + 2vyAδvy + 2vzAδvz A, A'が同一球面上 ⇒ vA = vA' ⇒一般化: 2vxδvx + 2vyδvy + 2vzδvz = 0 定数λをかけ、(1)式の辺々を加える [f'(vx)/f(vx) + 2δvx]δvx + [f'(vy)/f(vy) + 2δvy]δvy + [f'(vz)/f(vz) + 2δvz]δvz = 0⇒ 恒等式 → 各項の値0となる ∴ f'(vx)/f(vx) + 2δvx = 0, f'(vy)/f(vy) + 2δvy = 0, f'(vz)/f(vz) + 2δvz = 0⇒ f'(vx) = -2δvx·f(vx) これはある関数を微分したものはもとの関数に-2λvxをかけたものになる。従ってこの関数はeの関数になるf(vx) = αe–λvx2, f(vy) = αe–λvy2, f(vz) = αe–λvz2 (α, λ: constant), α = √(λ/α) ⇒ Def. Maxwell-Boltzmann分布則: 液体・気体の運動様式を表わせるマックスウェル・ボルツマンの速度関数: 気体の速度分布式 → 温度が高くなると広く分布 n(U) = N(m/2πkBT)3/2 … (1: マックスウェル式)exp[mv2/2kBT] 速度vで積分したものが右式 n(E) = N(e–E/kBT/Σe–E/kBT) … (2: ボルツマン式)Pr._dnx = nf(vx)dvx … (1) f(vx) = αe–λvx2 … (2) (1)に(2)を代入し、(1)の左辺を一般化し両辺を積分:
∫–∞∞nαe–λvx2dvx = ∫dnx = ∫dn = n (1)と(3)式を下式に代入
dn = nf(vx)f(vy)f(vz)dvxdvydvz = n(λ/π)3/2e-λv2dvxdvydvz
= 4π(λ/π)3/2·{(3√π)/8λ5/2}= 3/2λ__Cf. ∫0∞xne–ax2dx
〈v2〉 = 3/m·kt = 3/2λ ∵ λ = m/2kT ボルツマン統計N = Σni, W = N!/(Σni) → logW = NlogN – N – Σi(nilogni – ni)Max(ni) = Max(logW) ⇒ logW = 0 Σδni = 0, Σδniεi = 0 ni = Ce–βεi ⇒ N{e–εi/RT/Σe–ε/RT} → 未定係数法 運動エネルギー, W: 各桝目に入る量がどのように決まるか (確立論的組合せ) van der Waals状態方程式 van der Waals equation of stateDef. 分子間力(ファンデルワールス力): 分子同士の引力理想気体の状態方程式 pv = RT [問題] [仮定1] 気体全体積に比べ気体分子自身の体積が無視出来る → 実在気体で不成立
体積vのうちb(気体分子1 molの体積)は気体分子で占められる → 気体分子の運動に有効な空間は(v – b)
b* = NA16πr3/3 = 4πr3NAφ/3 = 4v, b*: 気体1 molの値 ⇒ この2点を補正 ⇒ van der Waals状態方程式 → 広範囲で実験値と一致 Van der Waals状態方程式: (p + a/v2)(v - b) = RT or (p + an2/v2)(v – nb) = nRT
a: 気体分子相互作用に関する物質定数 気体gasの液化 liquefactionDef. 臨界温度 critical temperature, Tk:
これ以上の温度では液化liquefaction不可能という温度
⇒ 臨界圧(力) critical pressure (Pk) 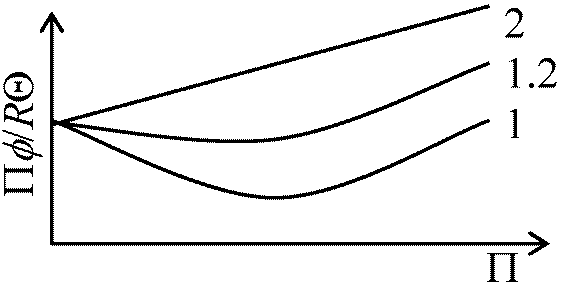 ⇒ 換算状態方程式 reduced equation of state:
⇒ 換算状態方程式 reduced equation of state:
(Π+ 3/φ2)(φ - 1/3) = 8/3·Θ [全気体に応用出来る式]圧力低-温度高: 理想気体に近づく → 状態方程式が利用出来る 圧力高-温度低: 状態方程式が当てはまらなくなる このPV = RTの補正をしたものがvan der Waals方程式と次のビリアル方程式 virial equation である PV = RT + B(T)/V + C(T)/V2 + D(T)/V3 + … B(T), C(T), D(T)は温度関数でビリアル係数と呼ばれる 気体の比熱気体1個の原子 E = 3/2·kT (三次元) ⇒ Tのみの関数ENA = 3/2·NA·kT = 3/2·RT (NA: アボガドロ数) T → 0, ENA → 0, energy → 0 ENAはその温度における1 molあたりの全エネルギーUである U = 3/2·RTCV = (∂U/∂T)V = 3/2·R Cp = CV + R = 5/2·R T: constant (∂U/∂V)T = 0, (∂U/∂P)T = 0一般に定圧・定容モル比熱の比 CP/CV = γ
1原子分子: γ = 3/5 熱力学の第2法則 second law of thermodynamics= エントロピー増大の法則Th. カルノー原理(定理): 熱 = 運動⇒ 第2法則(熱力学の第2法則 second law of thermodynamics) Def. 効率: 熱のうち仕事になった割合, η = W/Q = (Q1 – Q2)/Q1 = f(T1, T2)
(ただし証明法を間違えた) |
カルノーの考察 = 熱力学の原理的考察1. 熱機関と推力機関の類似作用及ぼす時: 高温 → 低温 (熱素 = 熱現象) [Cp. 高所 → 低所 Ex. 水] 2. 熱機関の特徴: 物体の形や体積の変化でも熱を運ぶa) 熱移動の2つの道筋 (熱伝導heat conduction)
高温 → 低温: 仕事をしない熱伝導 (熱の移動) heat transfer
水-熱素: 水力機関との類似性
高温熱源から低温熱源までの熱移動のゆっくりとした熱機関は準静的 Thomson: 熱機関は2熱源必要 = 「熱源からとった熱を仕事に変え、他に何の変化も残さない熱機関不可能」 Clausius: 「何らかの他の変化を残さずに熱は低温物体から高温物体へ移動できない」 自然界で起る変化
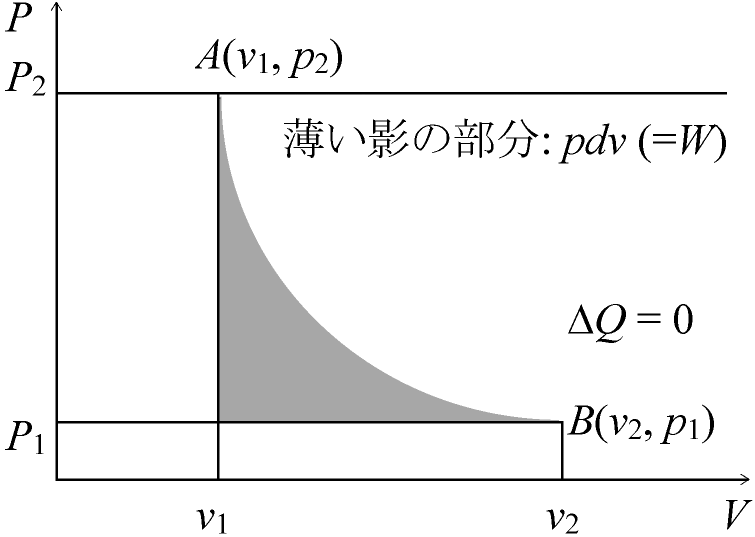
→ 可逆変化 reversible change + 不可逆変化 irreversible change ⇒ 仕事量energyに変化が起っているので不可逆変化 準静的過程 quasi-static process: P2 → P1, P1 → P2の過程Ex. ほとんど平衡を満たしたままピストンをdx動かせるような過程 可逆変化 reversible process: ハッチで示した範囲で仕事が起った場合、両過程でエネルギー変化がない準静的変化: 「(非常に)ゆっくりとした変化」= 常に熱平衡が成立していると仮定した変化
dw1 + dw2 = 0
線膨張率 coefficient of linear expansion
熱膨張 thermal expansion → 熱膨張率 coefficient of thermal expansion
大気圧と大気温度の高度変化1) T = constant = P(k)
dP = -p(h)dh gMh: 質量Mの物体がh m持ち上げられた時の位置エネルギー
P(h) = P0e-(Mg/RT)·h
dP = -ρgdh … (1) (1), (2), (3)より
P(h) = P0{1 – (1 – 1/γ)(gM/RT0)h}γ/(γ – 1) ≡ 熱を自発的に(= 仕事を熱に変換なしに)低熱源から高熱源に移動は不可能 ≡ 有限な装置を用い熱エネルギーを完全・無制限に仕事に変換は出来ない ≡ 孤立した系のエントロピーSは増大する ⇒ 第2種永久機関: 1つの熱源から熱をとり、その熱を全て仕事に変える熱機関
= 熱効率が1となる永久機関 (不可能, Ostwald) 可逆ならQを戻しWを取り出せる機関が存在 → 存在しない Def. 第1種永久機関: 力学的(熱的)エネルギー供給なしに無制限に仕事の出来る機関
= 閉ループ(閉じたループ) closed loopとなる
ΔV = Q – W, W = 0 ⇒ V = Q Δu: 1 molについてのエネルギー変化 b) 定圧変化 isobaric change
W = PΔv
Δu = Q - W = 0 ∴ Q = W
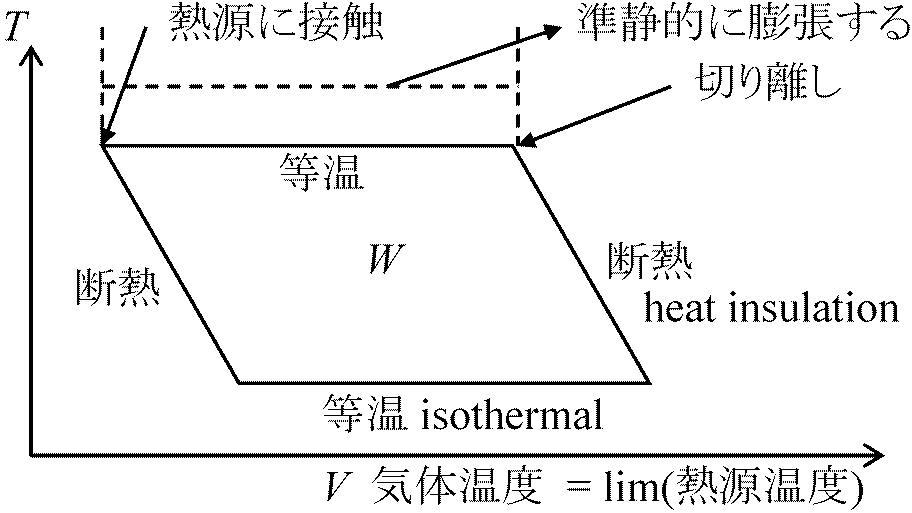
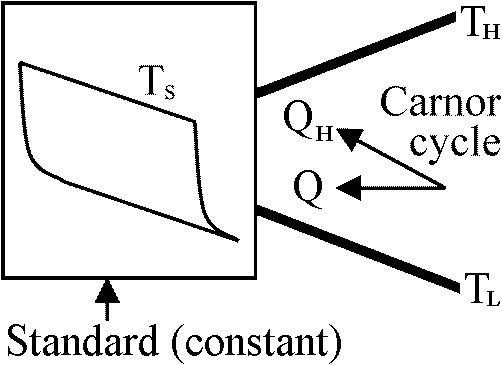
(∂u/∂T)T = Cv カルノーサイクル Calnot cycle (Calnot's cycle)熱の本性: 熱機関の構造
↓ 同じ熱素の移動(限りなく)  カルノー原理: 熱の動力(効率)は、それを取り出すために使われる作用物質にはよらない。その量(効率)は、熱素が最終的に移動しあう2つの物体の温度のみで決まる。但し、動力を発生させる方法は可能な限り完全に達しているものとする。換言すれば、温度差のある物体同士の接触が全く存在しない時である
カルノー原理: 熱の動力(効率)は、それを取り出すために使われる作用物質にはよらない。その量(効率)は、熱素が最終的に移動しあう2つの物体の温度のみで決まる。但し、動力を発生させる方法は可能な限り完全に達しているものとする。換言すれば、温度差のある物体同士の接触が全く存在しない時であるカルノーサイクル Carnot cycle 熱機関 thermal engine (heat engine) - Cyclic 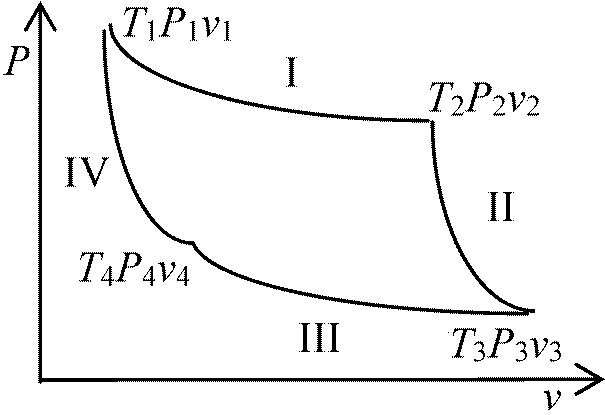
ΔU = Q - W I: 定温(等温)膨張 isothermal expansion 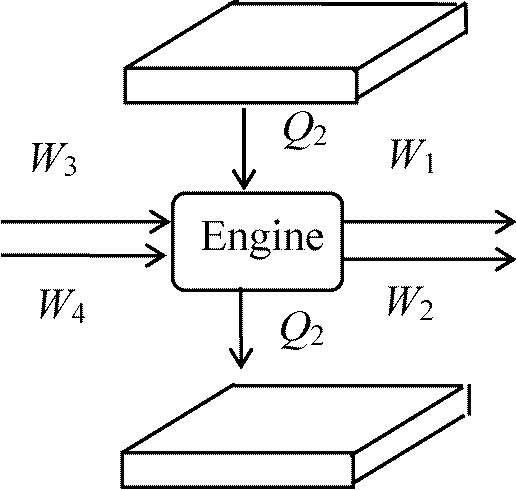
T1 = constant
Q = 0
ΔU = 0
Q = 0 ΔU = 0 Def. 熱機関効率, f
= (外界に対してした仕事)/(高熱源から吸収した熱量)
P2v2γ = p3v3γ, P1v1γ = p4v4γ ∴ P2/P1(v2/v1)γ = P3/P4(v3/v4)γ … (1)
|
|
一つの系 a system ⇒ p = 定数 constant, V = 変量 variable W = pΔV (外界に対する仕事) + W’(それ以外の仕事) = Q - ΔU ΔU + PΔV = Q - W (V: 状態量 ⇒ If 定圧 → pΔVも状態量) Def. エンタルピー enthalpy (熱含量 heat content), H ≡ U + PV定圧下で状態量 (気体のトータルの熱量とも言われる) ∴ ΔH = Q – W' (≠ 電池など電気的仕事を外に対して行う系)普通の系: 自己膨張Wのみ → W' = 0 ∴ ΔH = Q Def. h: 1 molあたりenthalpy → h = u + Pv理想気体 ⇒ Pv = PT ∴ h = u + RT Q.: 100°C, 1 atm: 1 mol water
→ 1 mol steam (水蒸発熱 = 40.681 KJ mol-1)
ΔU = ΔH - PΔV, ΔV = ΔVsteam – Vwater (Vwater ≈ 0) = Vsteam ≡ 1 mol溶液温度を1°C上げるのに必要なエネルギー C = 1 cal/g·degree (at 4°C) Ex. EtOH = 0.545 (0°C), Fe = 0.107 (0°C) Def. 熱容量 heat capacity, Cp: 系の温度 1°C↑ ⇒ 要する熱量= 比熱 × 質量 系 P = constant ΔH = QCp = (∂Q/∂T)P = (∂H/∂T)P 系 V = constant 閉鎖系 ⇒ W = 0 ∴ ΔU = Q - W = QCv = (∂Q/∂T)V = (∂U/∂T)V |
Def. モル比熱(モル熱容量) molar heat: 1 molでの熱容量 Def. 比容: 単位質量あたりの体積, α Def. 定圧熱容量: 圧力一定, CP, cp
ΔW = pΔV → ΔW = pΔα → ΔQ = Δu + pΔα (u: 1 molに対する全エネルギー) cp = (∂Q1 mol/∂T)p = (∂H/∂T)p, Cv = (∂Q1 mol/∂T)v = (∂u/∂T)v
h = u + RTをTについて微分 → (∂h/∂T)P = (∂u/∂T)V + R cは各気体によって決まる定数 (実験測定値) 表. 気体の比熱定数(273-1500 K)。a, b × 103 + c × 107
H2O = 30.36, 9.61, 11.8
H2 = 29.07, -0.836, 20.1
ΔQ = CpΔT – aΔp ⇒ Cp = Cv + R [Mayorの関係式]
Cp = 1004 JK-1kg-1 (定圧比熱) R: 抵抗, I: 電流, k: 比例定数 Def. Qj ≡ ジュール熱 Joule heat (電流から発生する熱) |
|
Def. 熱化学方程式 thermochemical equation A = B + X____Def. 反応熱 ≡ X, 熱量
定容条件 ⇒ 定容反応熱 ≡ Xv 物質状態は自明のとき以外は明記: H2O(s), H2O(l), H2O(g), 水溶液: aq Def. 発熱(吸熱)反応: X >(<) 0反応熱の種類 sort of reaction heatDef. 生成熱: 1 molの物質が構成元素の単体から出来るときの反応熱Def. 標準生成熱: 標準状態にある成分元素から出発し標準状態の化合物1 molを生ずる生成熱 標準状態 normal state ≡ 常温 25(or 20)°C, 常圧 1 atm
Ex. C(s) + 2H2 = CH4 + 75 kJ 1 cal = 4.1840 J (4.1840: 熱の仕事当量) Ex. ベンゼンの標準生成熱: Hessの法則より
(a) C (amorphous) + O2 (g) = CO2 (g) + 394.97 kJ 6C6H6 – 80.34 kJ [燃焼熱と生成熱は裏表] ともに1 mol (気体では0°C, 1 atm, 22.4 l)
生成熱を求める ⇒ 原系、生成系各物質の燃焼熱を得る必要 Def. 燃焼熱: 物質1 molが完全燃焼する時の反応熱 Ex. CH4(g) + 2O2(g) = CO2(g) + 2H2O(l) + 891 kJ Q. 0°C, 1 atm, 石炭ガス 1 m3 (水素50%, メタン30%, CO 10%, 窒素10%)を完全燃焼させた時の発熱量燃焼熱: 水素 68 kcal, メタン 213 kcal, CO 68 kcal A. 水素 500 l, メタン = 300 l, CO = 100 l (窒素 = 不燃気体)
発熱量: 水素 68 × 500/22.4, メタン 213 × 300/22.4, CO 68 × 100/22.4
→ 希薄強酸・強塩基水溶液の中和: 酸や塩基の種類によらず ≈ 56.5-57.5 kJ Ex. Na2SO4·10H2O + aq = Na2SO4 aq + 10H2O(l) + 78.5 kJ 熱化学方程式解法: 方程式作る → 未知数x → 只管解く! (組立法/消去法)Def. 結合切断エネルギー bond dissociation energy: 分子結合切断時に必要なエネルギー = 結合エネルギー Ex. (化学)結合エネルギー
C(graphite) + O2 → CO2(g)__________(1) ΔH1 = -395.5 KJ/mol ΔH: CH4生成エネルギー H2(gas) → 2H(atom) ΔH1 = 217.94 KJ/mol (ΔH1: 解離エネルギー)
C(graphite) → C(atom)___________ΔH2 = 715.0 KJ/mol |
20 min, 70 kg (weight)の人が150 mの高さまで階段を駈けあがると85.7 J/sの出力を要する
出力単位 J/s (Js-1) 馬力 hose power, hp: 1hp = 550 lbの物を1 sec.に1 feet上げる出力
1 lb ≈ 454 g = 0.545 kg, 1 feet ≈ 0.305 m
人間の筋肉 0.3 hp/kg → 70 kgの人の45%は筋肉 (= 9.4 hp) → O2のみ = 24 l O2/min
-C(OH)-H + O2 → CO2 + H2O + 500 kJ
反応熱は変化過程に依存しない
1) 直接生成: C(s) + O2 = CO2 + 394 kJ ⇒ CO + 1/2·O2 = CO2 + 283 kJ ⇒ (111 + 283 = 394) kJ Eq. カーチョフの式 Kirchoff's equation
反応熱が温度変化とともに変化する様子を与える式
hir, uirは原系のi番目物質の1 molあたりH, U
(1), (2)式を各々Tについて微分 [原系物質の比熱に+(左辺)、生成系物質の比熱に–(右辺)符号をつける約束]
(∂Xp/∂T)p = Σnicpi, (∂Xv/∂T)v = Σnicvi
H2 + 1/2·O2 = H2O →
= Σni[aiT + 1/2·biT + 1/3·ciT2]T0T
Σniai = (29.07 + 1/2 × 25.72 – 30.36) = 11.57 – 1/3 × 11.0 × 10-7{T – (298)3} JK-1mol-1 |
|
Def. 自由エネルギー free energy, Ef: 他エネルギー形態へ100%転換可能
= 常に仕事とし利用可(仕事になりうるエネルギー) Ex. 熱力学的特性関数 ⇒ F, G 生物体内で生じるエネルギーはEfでないと活動できない Ex. ATP Def. 束縛エネルギー bounded energy, Eb ≠ 自由エネルギーEx. 熱エネルギー thermal energy ↔ 仕事エネルギー Def. 全エネルギー(内部エネルギー) total or internal energy, U ≡ Ef + Eb★ 自由エネルギー free energy定温(等温)過程: f(T, V, P) → f'(T, V', P')〇ヘルムホルツの自由エネルギー: 定容(等積), V = 一定Helmholtz energy, F (or A) ≡ U - TS (TS 束縛エネルギー)
仕事とし取り出せる内部エネルギー量 ∵ ΔU = TΔS - PΔV ΔS ≥ ΔU/T ∴ ΔU - TΔS ≤ 0 (∵ U 一定 ⇒ ΔS ≥ 0. S 一定 ⇒ ΔU ≤ 0) ΔF ≤ ΔW
TΔS ≥ ΔQ (クラジウスの不等式). ΔU ≤ ΔQ + ΔW (熱力学第1法則) ⇒
最大仕事: 系になされる最大の仕事はΔFと等しい 〇ギブズの自由エネルギー: 定圧(等圧), P = 一定= 熱力学ポテンシャル thermodynamic potentialGibbs energy, G ≡ H - TS = U - TS + PV (∵ H = U + PV) = F + PV
Hの中で自発的変化で取り出せるエネルギーの最大値 系平衡状態 = 自発的変化起こらず(ΔG < 0)、G最小状態 |
ΔG = ΔF + PΔV (∵ G = F + PV)
If ΔV ≈ 0 (Ex. 固体・液体) → ΔF ≈ ΔG ΔU = ΔQ + ΔW (仕事 W)= ΔQ + ΔW' + ΔW'' (W = W' + W''. 膨張仕事 W'. 非膨張仕事 W'') W' = -PΔV (ΔS > Q/T ∴ TΔS > Q)
ΔU + ΔPΔV - TΔS ≤ ΔW'' A. T1 = 298 K, P2 = 1 atm, ΔS = Cpln(T2/T1) - Rln(P2/P1) = S° - RlnP → 温度一定・等温膨張 - エントロピー増大 断熱変化(Q = 0) ⇒ -W = -ΔU等温変化(T = 一定) ⇒ −W = −Δ(U − TS) = −ΔF Def. 化学ポテンシャル chemical potential, μ, ≡ g°: 1 molあたりのG (J/mol)
閉鎖系 ΔU = ΔQ + ΔW ⇒ 開放系に拡張 ΔQ = TΔS … (1) W = -PΔV … (2) ∴ ΔU - TΔS + PΔV = ΣiμiΔni … (3)
W = -PΔVと(3)式を(2)式に代入 ΔU - TΔS + PΔV = ΣiμiΔni
T = constant ⇒ TΔS = TΔS + SΔT = Δ(TS) Δ(U + PV - TS) = ΣiμiΔni H = U + PV. G = H - TS ⇒ Def. ΔG = ΣiμiΔni ⇒ μi = (∂G/∂ni)T,P,nj(j≠i) = G/n |
⇒ 単位
|
°C, °K: 冷温の尺度 Ex. 0°C, 1 atm: 氷融点(3重点) - 100°C, 1 atm: 水沸点 Def. 標準温度 standard temperature: (計量法で体積計の)容積を示す時に標準とする温度 (= 20°Cが多) 定点(温度の) fixed points ⇒ 温度目盛作成には任意の2点の定義定点必要 摂氏温度目盛 Celsius scale, °C 1742 Celsius, Anders (1701-1744): 摂氏温度の基礎考案
1 atm, 水融点(凝固点) = 0°C 温度: 以前は特定物質(理想気体、水銀熱膨張率等)の性質で定義 ⇒ Q. 水銀温度計・アルコール温度計: 細管中液体熱膨張利用 → 熱膨張率は温度により異なる バイメタル bimetal: 熱膨張率異なる2種金属板を合わせたもの
温度変化で熱膨張率の小さい金属の方に曲がる → 自動温度調節・自動スイッチ等、家庭電化製品に利用 |
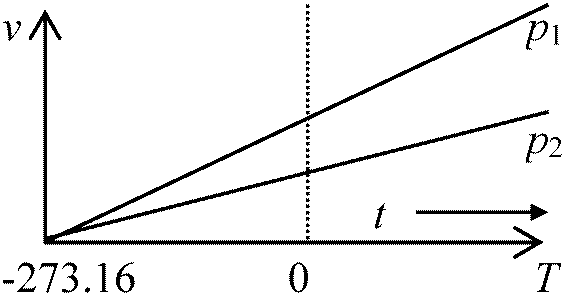
熱力学的温度(絶対温度absolute temperature, T)可逆熱機関の効率は作業物質に無関係(依存しない)ηrev = 1 – Q2/Q1 = f(Θ1, Θ2); Q2/Q1 = g(Θ1, Θ2) η1 = 1 – Q2/Q1, η2 = 1 – Q3/Q2, η3 = 1 – Q3/Q1 Q3/Q1 = f(Θ1, Θ3) = Q2/Q1 × Q3/Q2 = Q3/Q1 = g(Θ1, Θ2)·g(Θ2, Θ3) g(x, y) = y/xはこの関係を満たす Def. 熱力学的温度: Q2/Q1 = Θ2/Θ1 → T2/T1
目盛りの定義: 水の3重点 triple point; T1 = 273.16 K 国際基準 Kerbin (K): 理想気体(Ar, He)熱膨張から計算し絶対温度定めた Def. 絶対零度, T0: 温度低下 → (理論上)分子原子運動完全停止 → 分子原子運動停止 = 絶対零度以下は存在しない(温度に上限はない) 零点エネルギー zero point energy: 絶対零度での物質の分子が保持している運動エネルギー ケルビン温度 Kelvin scale (色温度 color temperature) : ケルビン温度目盛(kelvin, K ケルビン)
セ氏(°C)と絶対温度(K)の関係: |
| 温度計 thermometer | |
|---|---|
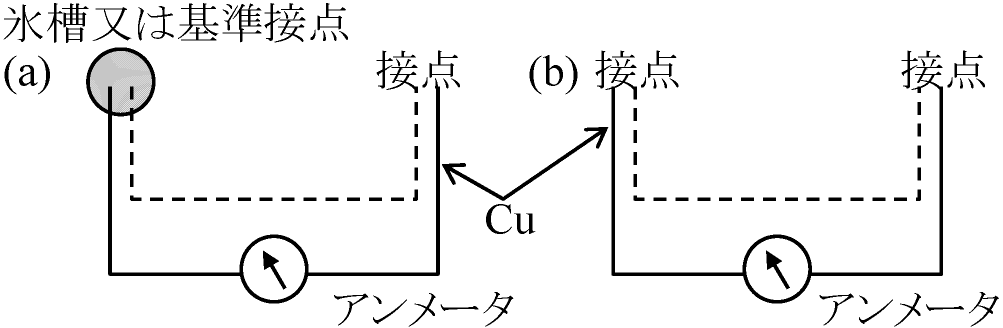
液体空気 liquid air (液体窒素 liquid nitrogen, 液体酸素 liquid oxygen) サーモグラフィ thermography 熱電対 thermocoupleQ ≡ 単位時間に発生する熱量トムスン効果: 場所により温度異なる導体(半導体)に電流通す → 導体内にジュール熱以外の熱発生(吸収) Q = gIaTI: 電流 aT: 温度差 g: 比例定数(トムスン係数または電気比熱) g: Cu, Zn > 0, Pt, Fe < 0, Pb ≈ 0 (熱起電力比較基準物質) ペルティエ係数 p、熱電能 n との間には、トムスンの関係式、g = dp/dT – n (成立が熱力学から導かれる)ペルティエ効果異種導体(半導体)接点に電流を通す → 接点でジュール熱以外に熱発生(吸収)が起こる現象 (Peltier Jean CA. 1785-1845, 仏)電流方向を逆にすれば熱発生と吸収は反対になる Q = PABI PAB: 比例計数(導体A, B間のペルティエ係数) |
自由電子が運ぶ熱量と電流の比が両導体で等しくないために起こる現象 →
比をそれぞれPA, PB ⇒ PAB = PA – PB
基準物質を同じとすれば、T(絶対温度)、熱電率 nA はPA = TnA関係が理論的に証明された ⇒ 熱電効果 thermoelectric effect (ゼーベック効果 Seebeck effect, Seebeck Thomas Johann 1770-1831 独) 起電力により回路に熱電流が流れる現象 ⇒ ペルティエ効果 + トムスン効果熱電対(熱電池) thermocouple, thermo-junction, themoelement熱起電力を利用した電流発生装置熱電帯: 多数の熱電対を直列につないだ装置 熱電対温度計熟電対を用いた温度差測定装置輻射エネルギー測定: 高感度必要
熱電対 → 輻射集中させる光学系必要 構造: 熱起電力利用 → 2種金属線を両端で接合 Ex. 銅-コンスタンタン、アルメル-クロメル、白金-白金ロジウム、ビスマス-銀 回路中に生ずる熱起電力の大きさ及び方向 = 両種金属の種類と両接合部の温度で決まる
赤外線が受光面にあたると接点温度↑ ⇒ 発生熱起電力を測定 [熱電図] (b) 両接点変温なら2接点間温度差計れる
0.1-0.2°C精度
利用: 土壌・空気・植物組織、表面温度、輻射熱等 |
|
クラジウス熱力学: カルノー原理肯定 = カルノー原理を如何に導くか = 熱に関与する現象中から、個々の物体や物質の熱的特性に関係のない、一般法則を取り出した理論 熱 = 運動 ∴ 熱 ≠ 保存 ⇒ この考え方からカルノー原理を導こうとする
QH' → → → → → QL'
超熱機関のした仕事(左辺) = カルノーサイクルの仕事(右辺)
エントロピー entropy時間とともに上昇 → 自己重力系で特徴的結論: カルノー・クラジウス: エントロピー増大の法則(第2法則)存在 (QH – QL)/QH = f(tH, tL) QL/QH = f(tH, tL) + 1 + g(tH, tL), Q0/Q1 = g(t1, t0), Q0/Q2 = g(t2, t0) ∴ Q1/Q2 = g(t2, t0)/g(t1, t0) ← T1/T2, Q1/T1 – Q2/T2 = 0t: その時の温度, T: 絶対温度 ⇒ TH, TLの2数の関数(温度関数)で表わされるlog[{t - (-273.12)/273.12}] 一考: 物理量の規定はある法則に基く → その法則がまた何に規定されるか理論の数学的表現カルノー機関: QH/TH - QL/TL = 0 通常機関: QH/TH - QL/TL < 0
状態量 (entropy): ΔS = ∫ABdQ/T ← 作業物質の受ける熱量 (Q'H – Q'L)/Q'H > (TH – TL)/TH → Q'H/TH – Q'L/TL > 0 系の外界に対する仕事 a. 定温可逆変化Q1W1, Q1W2ΔU = Q1 - W1 = Q2 - W2 … (1) ∴ Δu ⇒ 状態量 (1)よりQ1 - Q2 (熱量) = W1 - W2定温状態で熱の変化は有り得ない(W1 → W2) よってW1 → W2, W2 → W1 ∴ W1 = W2⇒ 定温可逆変化でWは状態量 ⇒ Qも状態量
p P2V1T3 P2V2T4 b. 一般可逆変化111 → 213 → 224 → … (1)111 → 122 → 224 → … (2) (1), (2)の2通りの道順によリW異なる ⇒
(1) W1 = P1(V2 - V1) 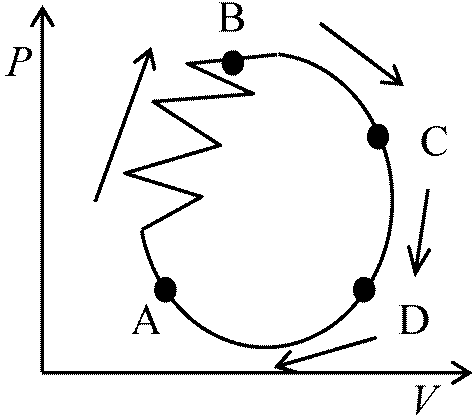 ∴ W1 - W2 = (P2 - P1)(V2 - V1)
∴ W1 - W2 = (P2 - P1)(V2 - V1)1 - (Q1 + Q2)/Q1 = 1 - (T1 - T2)/T1 ∴ -Q2/Q1 = T2/T1 or Q1/T1 + Q2/T2 = 0 閉鎖系における一般可逆変化により拡張 ∮(dQ/T) = 0 (1サイクルについての積分記号) Carnot cycle: Q1Q2 Q1/T1 + Q2/T2 = 0 ∴ Q1/T1 = -Q2/T2 ∮(dQ/T) = 0,∮ABC(dQ/T) + ∮CDA(dQ/T) = 0 ∮ABCA(dQ/T) + ∮ADA(dQ/T) = 0 D = ΔS (状態量変化) ⇒ Def. ΔS: エントロピー(秩序・無秩序の度合) = ∫(dQ/T) (可逆変化)  エントロピー, S ≡ Σi(Qi/Ti) = ∫(dQ/T) [状態量]
エントロピー, S ≡ Σi(Qi/Ti) = ∫(dQ/T) [状態量]Law. エントロピー増大の法則 Pr. Case. 可逆過程: 状態のみでA ⇔ Bエントロピー決定
→ 不可逆過程でも同様に考えられる
⇒ ∫S1(A, B)(dQ/T) = –∫S2(A, B)(dQ/T) ∴ ΔS = -ΣiQi/Ti > 0 ⇒ Cp. 不可逆変化定温可逆変化: T = constant, Q/T = ΔS, Q = TΔS 外界から系になされた仕事 W ⇒ ΔU = Q - W = TΔS - W 定温不可逆変化: T = constantWi = W → [irreversible] → ΔU + W → ΔU + Wi < TΔS → Qi < TΔS or Qi/T < ΔS ΔS ≥ Q/T: 等号は可逆変化のとき成立 エントロピー増大の法則 law of entropy
※ 全宇宙のエネルギー一定 ⇒ 全宇宙のエントロピー増加し続ける 孤立系 → ΔS > Q/TQ = 0∴ ΔS ≥ 0 ⇒ 可逆変化 → ΔS = 0, 不可逆変化 → ΔS > 0 微小な断熱、定温変化 ΔS ≥ Q/T → ΔS ≥ S·dQ/T … 有限 dS ≥ dQ/T … 微小 熱力学の第二法則: 高い熱源から低い熱源にのみ熱の移動が起こる 孤立系: Q = 0, ΔS = 0
T1 (高) → [ΔQ] → T2 (低) 2) ΔQ/T2 > ΔQ/T1 ⇒ 1/T2 > 1/T1 ∴ T1 > T2 熱力学公式: P, V, U, H, T, S, F, Gの関係H = U + PV G = F + PV F = U - TS G = H - TS
過程 全エネルギー 自由エネルギー
ΔU 定容反応熱 dU = TdS - PdV H = U + PV → 両辺微分 ⇒ dH = dU + PdV + VdP F = U – T → 両辺微分 ⇒ dF = dU - TdS - SdT
dU = TdS - PdV
dH = TdS + VdP
また dU = TdS – PdV ∴ K = T, L = -P, y = S, z = V
(∂X/∂U)z = (∂X/∂y)z(∂y/∂U)z ∴ (∂X/∂y)z = (∂X/∂U)z/(∂y/∂U)z
(∂y/∂z)x = -(∂X/∂z)y/(∂X/∂y)z
dU = TdS - PdV, (∂T/∂V)S = -(∂P/∂S)V [ ° = 1 mol ] 理想気体 ideal gas (perfect gas)U° = U° + CVT (U°: 全エネルギー U°0: T = 0の時の全エネルギー)H° = H°0 + CpT (H°: 全エンタルピー H°0: T = 0の時の全エンタルピー) dS = (dU + PdV)/T S° = S°0 + ∫T0TdU/T + ∫V0VPdV/T (S°0: T = T0におけるエントロピー) PV = RT ⇒ S° = S°0 + Cvln(T/T0) + Rln(v/V0) (T0 K, V = 1 l) S° = S°0v + RlnV (S°0V: エントロピー) dS = (dH - VdP)/T
S = S0 + Cpln(T/T0) - Rln(P/P0) |
S°0P: TK, P = 1 atmの時のエントロピー F° = U - TS (F: Helmholtz energy)U = U0 + CVT S = S0V + RlnV F° = U0 + CVT - TS0V - RTlnVF° = F0V - RTlnV (F0V: TK, V = 1 (l)の状態でのfree energy) U = U0 + CVT S = S0P - RlnP F° = U0 + CVT - TS0P + RTlnPF° = F0P + RTlnP (F°0P: TK, P = 1 atmのもとでのfree energy) Th. 溶液反応: 混合前に各成分が混合後に示すべき分圧にしておけば混合による仕事がない Pr. T = constant, V1: gas 1, n1 mol. V2: gas 2, n2 mol ⇒ V = V1 + V2 g1 = g01 + RTlnp1 g2 = g02 + RTlnp2 理想気体概念を導入: ΔV = 0, ΔS = 0S' = Σnisi' = Σnis01p - Rnilnpi or S' = ΣniP0iP - RΣnilnP - RΣnilnxi S°i0: 外圧Pのもとで単独に存在する場合の1 molでのS ⇒ 総和
S° = ΣniSi0
混合後のHelmholtz's free energy (F')は Gi0 ≡ μi0 μi = μi0 + RTlnxi 
液体と固体蒸発曲線: 1成分2相van der Waals状態方程式: 液化から始まる気体-液体共存状態 ⇒ ある1温度で気体gas (蒸気vapor)圧力一定 飽和状態: 各層への各々の移動する分子量が等しい 気体の液化
n: mol number pV = n2RT … (3) (2)に(3), (1)を入れる P = (W/M·P)/(W/M + PV/RT) … (4) ⇒ (4)式によって得られた曲線 ≡ 蒸発曲線 (水)蒸気圧 (water) vapor pressure ⇔ 大気圧 ⇒ Def. 沸騰 ebullition (boiling): この時に気泡を生じた状態外圧一定になり沸点boiling pointも従って一定: 1 atm = 760 mmHg(標準) Clapeyron-Clausius式蒸発熱(気化熱) heat of evaporation: 液体分子が蒸発するには液体内分子間引力に打ち勝たなければならない。そのために必要なエネルギーは外から熱エネルギーとして供給される⇒ 一定温度で一定量の液体が蒸発するのに必要な熱量のこと Def. モル蒸発熱: 1 molあたりの蒸発熱 Clapayron-Clausius式 dp/dT = LV/T(vg - vl) … (1)
dp/dT: 温度Tにおける蒸発曲線の勾配 dP/dT = LVP/RT2 … (2) dP/P = LV/R·dT/T2 … (3) LV = constantと仮定し両辺積分 lnP = -LV/RT + C … (4)logP = -LV/2.303RT + C … (5) → logPを1/Tに対してプロットしたものが直線となる log(P2/P1) = LV/2.303R·(T2 - T1)/T1T2 … (6) ⇒ 曲線の任意の点における勾配に-Rを乗じたものが蒸発熱となることを示す Law: Troutonの法則 LV/Tb ≈ 21 calK-1mol-1 = 92 JK-1mol-1, Tb: 沸点(K), LV: mol蒸発熱
無極性溶媒で良く当てはまり、蒸発エントロピーがこの種の全液体でほぼ同じであることを示す 蒸気と液体の化学ポテンシャルが互いに等しくなくてはならない G = nlμl + ngμg
μl: 液体の化学ポテンシャル G' = (nl + δn)μl + (ng - δn)μg 移動前後の系のGibbs's free energyの変化量(ΔG)は ΔG = G' - G = δn(μl – μg) 平衡条件 ⇒ ΔG = 0 , dG/dv = 0 … (8) ∴ μl - μg = 0 or μl = μg ∴ μl = μg = μ0g + RTlnP or RTlnP = μl - μg … (9) G = U - TS + PV = F + PV = H – TS … (10) この式を1 molあたりになおすと: μl = h0g - Ts0g, μg = hl - Tsl … (11) (11)式を(9)式に代入 ⇒ lnP = -(g0g - hl)/RT + (s0g - sl)/R … (12) h0g - hlはp = 1 atmにおける蒸気のエンタルピーと液体のエンタルピーの差。よってmol蒸発熱に等しい ∴ h0g - hl = LV … (13) (s0g - sl)/R = C = const これと(12), (13)式から(4)式が導ける Troutonの法則では(11)式より μ0g - μl = (h0g - hl) - T(s0g - sl) … (14) T = TbにTをとる(Tb: 沸点) μ0g = μg - μl … (15) よって(14)式は h0g - hl = Tb(s0g - sl) (13)式を用いて LV/Tb = s0g - sl … (16) ⇒ Troutonの法則: 蒸発エントロピーが各物質で同じ値を持つことを示す [経済学的定義] エネルギーと資源エネルギー変換 energy conversionエクセルギ(有効エネルギー) exergy: 機械的仕事に変換できる ↔ Def. アネルギー(無効エネルギー) anergy: 変換できない エクセルギ: エネルギーは量的に保存されても質的には高位から低位に変化
→ エントロピーが不可逆的に増す過程を表す量 = 省エネルギーで節約すべき量
⇔ 自然界ではエネルギー変化は殆どが不可逆的
Q' ↖ ↗ W' W
閉鎖系のエクセルギー = U – U0 – T0(S – S0) – P0(V0 – V) Ex. 熱伝導損失 → 供給電気エネルギー
発電所効率 Max ≈ 40% Ex. 薪、石油、石炭、天然ガス、水力、地熱、風力、太陽熱・光、原子力 Def. 2次エネルギー資源secondary energy source: 1次エネルギーを工業的に加工・転換し作られる資源Ex. 電気、ガソリンや灯油等の石油製品、製鉄用コークスや都市ガス等の石炭製品 Def. 再生(更新)不能エネルギー資源nonrenewable energy resource: 1度使うと永久になくなるエネルギー資源Ex. オイルシェールやオイルサンド等の石油類似燃料を含む合成燃料の原料資源、石炭液化・ガス化による合成燃料(合成石油・合成ガス、メタノール) Def. 再生可能エネルギー資源(更新できる資源renewable resource): 自然力から繰り返し利用できる資源
|
|
Def. 相 phase (of matter): 物理的化学的性質が均質部分
Ex. 気相・液相・固相(結晶相crystal phase: 結晶構造差) Def. 不均質(不均一) heterogeneous: 2つ以上の相から出来ている系 Def. 遷移温度 transition temperature: 物質がある温度を境として破壊形態が変わる変曲点の温度 Def. 相転移 phase transition (change): 相が変わること Ex. melting, boiling, transition → エネルギー(熱)必要 ⇒ 相転移に伴なう変化 = 蒸発熱、V、H: 物質が与えられた条件(T, P, …)下でどのような状態(相)をとるか状態を決める2要素 ⇒ エントロピーの高い状態をとろうとする
1) エネルギーの低い秩序構造をとろうとする傾向
温度上昇に使われず隠れてしまった熱 (= 次の相の構造に蓄えた) Def. 転移温度(点), T(m) = dH/dS (ΔG = ΔH - T(m)ΔS = 0) [全系で成立] 固体 → 液体: 融解 melting
融点(融解点) melting point - 融解熱 heat of fusion
= 凝結・凝縮 condensation
気化熱(蒸発熱) heat of vaporization - 沸点 boiling point 固体 ↔ 気体: 昇華 sumlimation 昇華熱 heat of sublimation 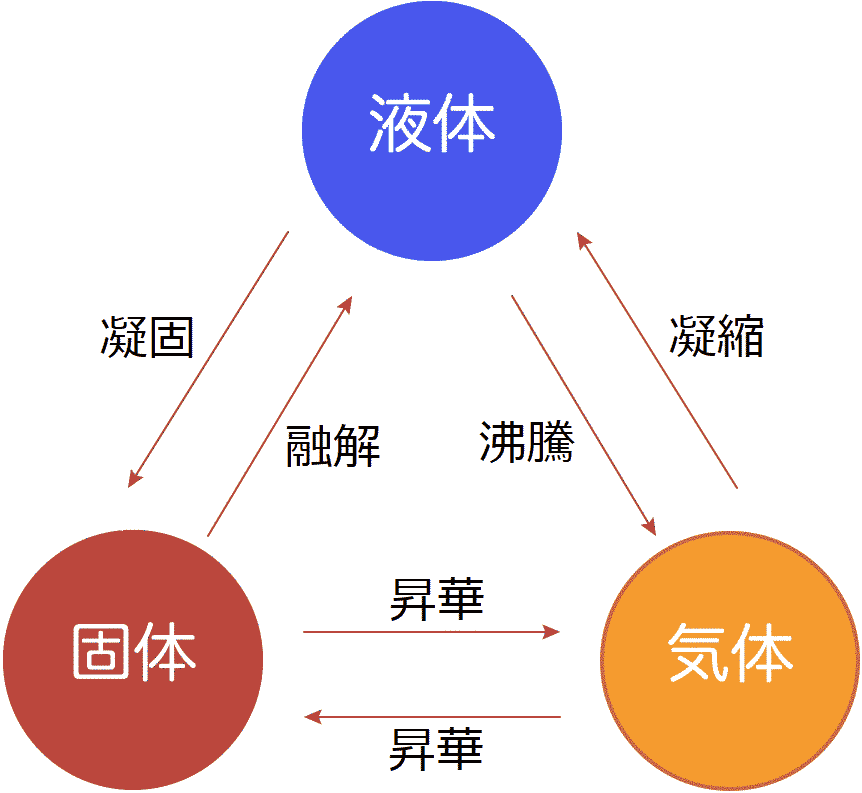
ヒートポンプ heat pump (heat mover): 気化熱利用し周囲物体冷却、 または凝固熱利用し加熱する装置
冷媒: この際の気体や液体 媒質中に置く高温固体の冷却に関する法則 ⇒ 固体から媒質への熱伝導速度 ∝ 固体表面積, ∝ 固体-媒質温度差 ⇒ -dQ/dt = αS(T – Tm) Q: 固体の熱量, t: 時刻, S: 固体表面積, T: 固体温度, Tm: 媒質温度 Def. 表面熱伝導率, a: 固体表年及び媒質の性質により決まる比例定数Def. 成分 component: 一相の組成を表す最小限必要な化学的に異なる成分 [c: 成分数 number of components] Def. 平衡状態 equilibrium state: 正反応速度 = 逆反応速度
⇒ 濃度(圧力)変化しない (平衡点 equilibrium position)
p: 平衡にある相の数 number of phases
平衡条件を用いる
xji = nji/Σjnji … (3)
理想気体: μji = μ0,ji + RTlnpji = μ0,ji + RTlnP + RTlnxji … (6) 2相 double phase: 蒸気 + 水(1成分2相系), 水 + 氷(1成分2相系) 3相 triple phase: 蒸気 + 水 + 氷(1成分3相系), gas + benzene + 水(2成分3相系) 一成分系 one component system⇒ 一成分(系)状態図 phase diagram相律 f = c – p + 2より
p_f_系数 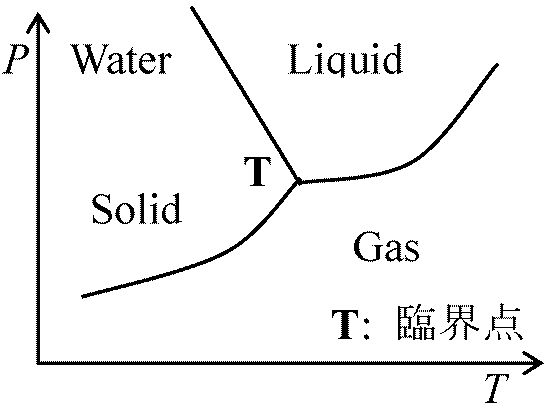 相転移のある物質
相転移のある物質
Ex. 斜方硫黄SI, 単斜硫黄SII → 複数の三重点 順安定 metastable: 昇華、融解、蒸発曲線→ いずれも明確な曲線 過冷却 supercooling: 物質が凝固点以下でも液体状態= 過冷却液体 supercooled liquid 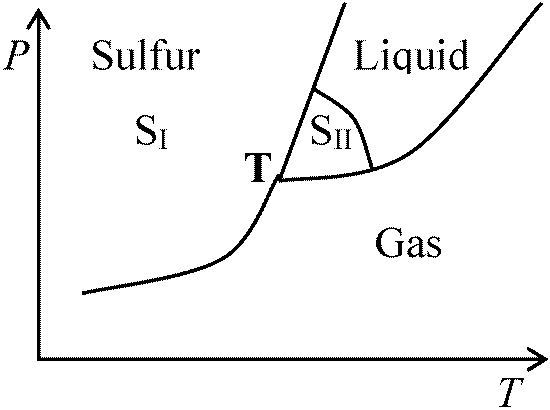 平衡移動のルシャトリエの原理: (一般に)平衡成立状態の条件を変化 ⇒ 影響を打ち消す方向に平衡が移動
平衡移動のルシャトリエの原理: (一般に)平衡成立状態の条件を変化 ⇒ 影響を打ち消す方向に平衡が移動
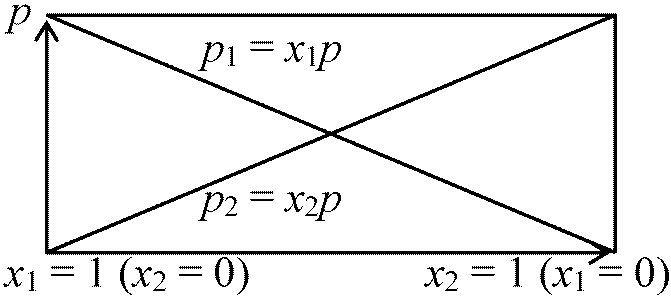
Ex. N2O4 = 2NO2 – 54.8 kJ (V = constant) ⇒ T↑ → NO2↑, N2O4↓ [温度低下] 理想溶液 ideal liquidμji = μ0,ji + RTlnpji = μ0,ji + RTlnP + RTlnxji
→ μA = μ0A + RTlnP + RTlnxA … (1) μA = μ0A + RTlnP … (2) ⇒ Def. 理想溶液: この式を満足する溶液 1) xAが0-1の範囲で(2)式成立 ⇒ 理想溶液(または完全溶液)2) xAが十分小さいとき成立 ⇒ 理想希薄溶液
m–A0 ≠ mA0 ⇒ 固相と溶液相の成分Xが平衡状態 [溶解度 ⇒ 飽和溶液量を考える] 結晶析出 = 飽和溶液
飽和溶液量/析出量 = (100 + 溶解度)/溶解度差 μxl (溶液中の化学ポテンシャル) = μxs (固体の化学ポテンシャル) ⇒ μxs = μxl = μxo + RTln(XA)sat … (1) (XA)sat: 飽和溶液 saturated solution中における溶質のモル分率 ↔ 不飽和溶液 unsaturated solution
ln(XA)sat = (μAs – m–A0)/RT … (2) ΔhA: Aの融解エンタルピー変化 (= 融解熱)
LV/Tb = s0g - slより ΔSA: 融解のエントロピー, Tm: Aの凝固点、融点
(6)を(5)に代入 ⇒ 2) 融解熱ΔhAがほぼ等しい物質 →
Tm (融点)低いものほど溶解度大(理想溶液のみ当てはまる。水溶液ではあまり当てはまらない) 溶液の蒸気圧
成分 モル 蒸気圧 純粋での pA = xApA0, pB = xBpB0 … (2) ≡ ラウールの法則 Rault's law xA: 0-1で成立は理想溶液のみ ↔ 一般に分子間相互作用のため不成立 成分が不揮発性であれば p0 = 0 → pB0 := 0 ↔ 揮発性液体 volatile fluid p = xApA0 or (pA0 - p)/pA0 = pA0(1 - xA)/pA0 = xB … (3) ≡ Raultの法則(拡張) |
Def. 蒸気圧降下 vapor-pressure depression: 液体に不揮発性溶質を溶解
→ 純粋の飽和蒸気圧より低下する ⇒ Law. ラウールの法則 Raoult's law: 相対的蒸気圧降下度 = 溶質モル分率理想希薄溶液 nA (溶媒物質量) ≫ nB (溶質物質量) ⇒ (pA0 - p)pA0 = nB(nA + nB) ≈ nB/nA … (4)
成分A: 溶質の質量 WA, 分子量 MA nB/nA = nB·MA/WA = m/1000·MA … (5) ∴ (pA0 - p)/pA0 = m/1000·MA … (6) Pr. 熱力学的Raultの法則の証明 μAg = μ0,Ag + RTlnpA pA: この蒸気におけるAの分圧
μAl = μA0,l + RTlnxAl Def. 過剰化学ポテンシャル excess chemical potential, μAE: その修正の際加える値 μA = μA0 + RTlnxA + μAE … (10) Def. fA: 活量係数 activity coefficient
lnfA = μAE/RT … (11) Def. xAfA: 活量 acitivity
理想溶液のxAをxAfAにし、非理想溶液に理論を拡張可能 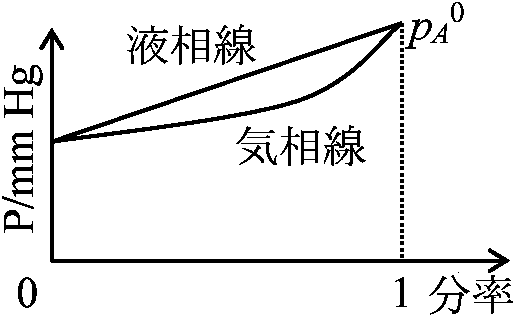
液相-気相平衡気体の溶解度pA = pxAg … (1) Dalton分圧則を仮定した形 (1)にp = pA + pB = pA0xAfA + pB0(1 - xA)fBのxAをxAlとして代入 xAg/xAl = pA0/p … (2)p (蒸気圧) < pA0 → xAg > xAl ⇒ 蒸気圧pと平衡にある液体ではA成分含有量が蒸気中より小さい Law. ヘンリーの法則 Henry's law: 一定温度 → 一定量 (ml) の溶液中w = kp
w: 一定量の液体への溶解量
⇒ 溶解する気体の質量 (g) はその気体の分圧に比例 A中にgas Bが溶けている時 ⇒ pA = xApA0, pB = kxB0 Ex. 0°C, 1 atm, O2 → 6.8 × 10-2 g/l(H2O)溶ける (酸素分圧 1/5)
1 atm → 6.8 × 10-2 × 1/5 g pB = k'c, c: 溶液濃度 Law. 分配の法則: 「互いに混ざり合わずに互いに接する2種の溶媒に1つの溶質を溶かし平衡になった時には、一定温度のもとでは各相における溶質の濃度の比は一定」
溶質Bのα及びβにおけるmol分率をxBα, xBβ、濃度をcBα, cBβとすると Def. 分配係数 ≡ KD or KD' Pr. ヘンリーの法則・分配の法則の証明(熱力学的)
μBl = m–B0,l + RTlnxBl … (1) lnpB = (m–B0,l - μ0,Bg) + lnxBl … (3)
m–B0,l- μ0,Bg = lnKB … (4) 分子量 MA, 密度 ρ, B成分mol濃度 cB
xB = nB/(nA + nB) ≈ nB/nA = cBMA/(1000ρA) を(4')式に代入
ΔTb: モル沸点上昇温度差, Kb: モル沸点上昇定数, m: 分子量 ΔTb = RTb2/LV·m/1000·MA = RTb2/1000lv·m lv: 溶媒1 gあたり蒸発熱 Kb = RTb2/1000lv Def. 凝固点降下: ΔTf = Kfm
ΔTf: モル凝固点降下温度差, Kf: モル凝固点降下定数, m: 分子量 浸透圧溶媒分子を移動させる力の大きさLaw. ファントホッフの浸透圧の法則: 体積Vの溶液中に溶質がn含まれるときの溶液の浸透圧πとの関係 ⇒ ファントホッフ式 van't Hoff equation, 体積V (l), 溶質 n (mol), 浸透圧 π (atm) πV = nRT, or π = CRT R: 気体定数, T: 溶液の絶対温度, C: 溶液の容量モル濃度 → C = n/V Pr. van't Hoff式の熱力学的導き方
π = pII - pI … (5) -ln(1 - xB) = xB + 1/2xB2 + 1/3xB3 + …
xB ≪ 1よってxB ≈ nB/nA その他の平衡固相-気相平衡L = R·(P/P)/(dT/T2) = R·dlnP/(-d(1/T))L: 分子昇華率, P: solid vapor pressure dlnP/(d(1/T)) = -L/RlnP = -L/R·1/T + constant 固相-液相平衡共融点 共晶 冷却曲線 寒剤 熱分析 高融点dP/dT(m) = L/T(Vl - Vs) T(m): melting point, L: 融解熱, Vl: liquid mol vol, Vs: solid mol vol dT(m)/dP = T(m)(Vl - Vs)/Lρ: density, M: 分子量 Vl = M/ρl, Vs = M/ρs∴ dT(m)/dP = T(m)M/L·(1/ρl - 1/ρs) > 0: 通常の物質, < 0: H2O (ρs < ρl) 固相-固相平衡L = T·dP/dT·(Vl - Vs) L: 転移熱凍結乾燥 freez dry (dehydration)真空凍結乾燥 vacuum-freez dry生物(植物)を凍結状態で水昇華させ乾燥 → ビタミン等成分維持 + 低タンパク質変性 + 色・香保持 基本的に水分を一旦氷結晶(純水)とし凍結させ4.6 mmHG (6.12 hPa)以下の真空条件下でエネルギー加え、昇華により氷結晶を水蒸気に変え除去 表. 高温乾燥法と凍結乾燥法の比較 水分除去方法: 凍結乾燥 = 氷のまま、融解することなく、昇華除去 通常乾燥 = 表面からの蒸発により除去。毛管現象により孔隙通り移動 形状・変化: 凍結乾燥 = 形状変化少ない通常乾燥 = 乾燥中の水移動 → 発泡/分離/濃縮/収縮/表面硬化等起こる 組織変化: 凍結乾燥 = 低温乾燥進行のため成分変化は殆どない通常乾燥 = 温度大、水分活性大により変質大 最終水分: 凍結乾燥 = 多孔質に乾燥 → 内部も乾燥し低水分通常乾燥 = 表層に濃縮層でき、深部乾燥困難 復元性: 凍結乾燥 = 多孔質となるため復元性・溶解性良い通常乾燥 = 収縮起こり、復元困難となる部分残る |
|
Def. 無熱溶液 athermal solution ⇒ 仮想溶液 = 放熱・吸収無視
高分子溶液概念導入のため 理想溶液の混合エントロピー式: ΔSH = -k(n1lnN1 + n2lnN2)N1 = n1/(n1 + n2), N2 = n2/(n1 + n2) ≠ 高分子溶液 → 高分子直径は低分子の100万倍に相当はよくあるコロイド colloid= ミセルが集合した状態ミセル micelle: 親水基と疎水基の両方をもつコロイド粒子の集合 コロイド状態: 顕微鏡では粒子見えない - 低分子より大きな粒子とし物質が分散した状態≈ 濾紙通過するが、半透膜通過できない大きさ Def. 凝集(力) cohesion: 安定性失った粒子(コロイド等)が寄り集まり塊になるEx. 塩化鉄(III)飽和溶液(黄褐色) + 沸騰水 → 水酸化鉄(III)
FeCl3 + 3H2O → Fe(OH)3 + 3HCl 沈殿 コロイド粒子 イオン等 1粒子中原子数(個) 109 10-5~10-7 103~1 直径(球状仮定, cm) 10-5 10-5~10-7 10-7 < 例 塩化銀 泥・牛乳 NaCl, ショ糖 分散質/分散相 dispersion phase: コロイドを作る物質 分散媒 dispersion medium: コロイドが分散している物質 土壌コロイド系: 分散相 = 固相 ↔ 分散媒 = 液相 → 2相不均質系 懸濁液 suspension: 比較的大きな粒子が液体中に分散 Ex. 粗粘土コロイド溶液 colloidal solution ゲル gel: 分散媒-分散質分離せず均一な固体 Ex. 寒天、ゼリー、ゆで卵
乾燥ゲル: ゲルが水分失ったもの Ex. 乾燥食品、髪 ヒドロゾル hydrosol: 分散媒 = 水 陰性・陽性コロイド粒子は帯電 ⇒陽性コロイド positive colloid or basoid = 電荷正 |
陰性コロイド negative colloid or acidoid = 電荷負
Pt, Ag, Cd, S, Se, I2のゾル 土壌: 両性コロイドと陰性コロイドの複合体 表. コロイドの分類
形状
集合状態
分散媒
分散質
例 ガラス繊維 fiberglass: 無機ガラスを高温で融かし繊維状に加工したもの コロイド水溶液中のコロイド粒子の種類水分子との結合状態
コロイドの特徴
|
|
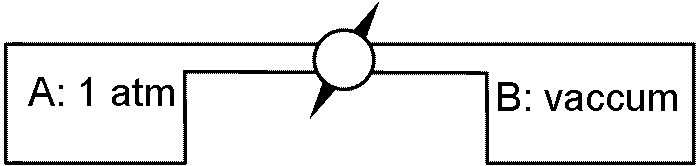
(1)理想気体をV0から2V0まで断熱自由膨張させる → 問に答えよ (a) 膨張の前後における内部エネルギーの変化 (b) 膨張の前後における温度の変化 (c) T-V図のうえに始めの状態(A)と終わりの状態(B)を示せ (d) 膨張の前後におけるエントロピー変化 (e) 膨張の前後におけるヘルム・ホルツの自由エネルギーの変化 |
(2) 上問で、(断熱自由膨張ではなく、)断熱的にゆっくりと膨張させる場合はどうか。上問(a)-(e)にの問に答えよ (3)エントロピーについて次の問に答えよ (a) 定積比熱Cvをエントロピー S(T, V)を用いて示せ (b) 体積一定ならば、Sは温度の単調増加関数であることを示せ (4)非可逆熱機関の熱効率(η')は可逆熱反応(η)より小さいことを示せ |


 PW1 + PW2 ≠ 0 ⇒ 不可逆
PW1 + PW2 ≠ 0 ⇒ 不可逆