(2020年3月3日更新) [ 日本語 | English ]
|
Def. 遺伝子 gene (☛ 遺伝学) 初期: ゲノムもしくは染色体の特定位置に占める遺伝単位 (≈ 遺伝子座) 現在 (s.l.) 塩基配列にコードされた遺伝情報 (s.s.) タンパク質一次構造に対応する転写産物(mRNA)情報もつ核酸分子上特定領域(= 構造遺伝子, シストロン cistron) |
シーケンス解析: 塩基配列を解析すること 塩基配列(DNA配列): DNAを構成する塩基(AGCT)の並び方 |
核酸研究 studies on nucleic acids1869 ミーシェル: 膿(白血球死骸)から核細胞分離(命名ヌクレイン nuclein) 高P(リン)含有 = 強酸性で高粘性 1871 ホッペザイラー & ミーシェル: red blood cell, yeast, yolk等同物質(ヌクレイン)取出す(他研究者達追試) + サケ白子からも発見 1872 ミーシェル: サケ白子(精子)
塩基性タンパク質(プロタミン)がnucleinと結合 - 発見 1889 アルトマン: ヌクレイン(核物質)を核酸 nucleic acid と呼ぶ → 普及 1901 コッセル: G, C, T, A, Uを核酸から分離し化学構造決定 → 核酸は2種類ある 1902 レベン: 酵母菌の核酸に含まれる5炭糖 = デオキシリボース1937, 38: フォイルゲン & カスペールソン DNAは核内のみに存在 (→ 否定) 1940's → RNAは植物細胞質・核内だけでなく、動物細胞細胞質も含むRNAがタンパク質合成に関与していることが強調される 1960年代(50年後半): 14C操作容易になる
→ 研究盛ん: 14CO2, 14C-amino acids中から得られた
Table. 種々細胞RNA, DNA, protein含量(RNA, DNA, protein含量合計を100%とした)
RNA(%)
DNA(%)
Protein(%)
RNA/DNA 核酸構成成分と組成分析= DNA + RNA= 塩基(AGCT/AGCU) + 糖 + リン酸 ☛ 核酸分析 核酸の化学修飾base, nucleoside, nucleotide, RNA(アルカリに弱い) ⇔ DNA(酸に弱い)ヌクレオシド nucleoside: N-glycoside bond
1) NMP: 比較的安定。NDP, NTP, cyclic NMP: 不安定, ATP ⇔ ADP + Pi |
実験系pH 5.5-8.5, Temp. 0-37°C, time: 短いほど良いBuffer: 制限多 Ex. アミンbufferはアルキル化に、カルボン酸・リン酸bufferはエステル化に使えない 試薬: 制限多 = 水に溶け分解されないものi) アルキル化 alkylation: アルキル基を転移する反応 → 核酸塩基の人為的化学修飾に使わる
メチルメタンスルフォン酸、エチルメタンスルフォン酸などがよく使われる
U, C → [Br2] → 5-Br*
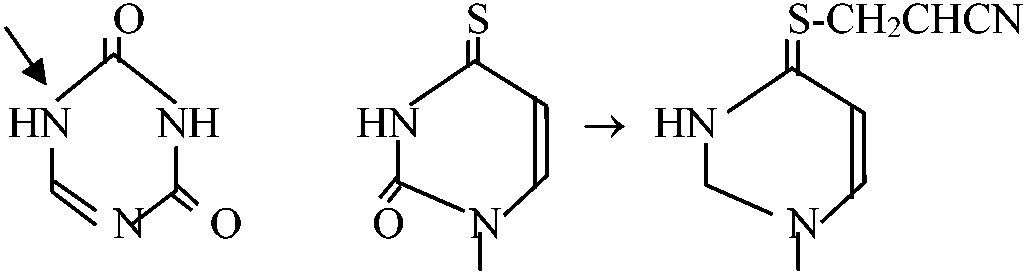
CH2=CH-CN, pH 11.5 - all reaction, pH 8.8 - none
(Canellakis et al. 1959) ヌクレオチドのde novo生合成ピリミジン pyrimidineはプレハブ工法、プリンpurineの方は在来工法によって生合成されるプリンヌクレオチド purine nucleotides: PPiの存在 → 生合成系の阻害/生体に対しては有利な機構 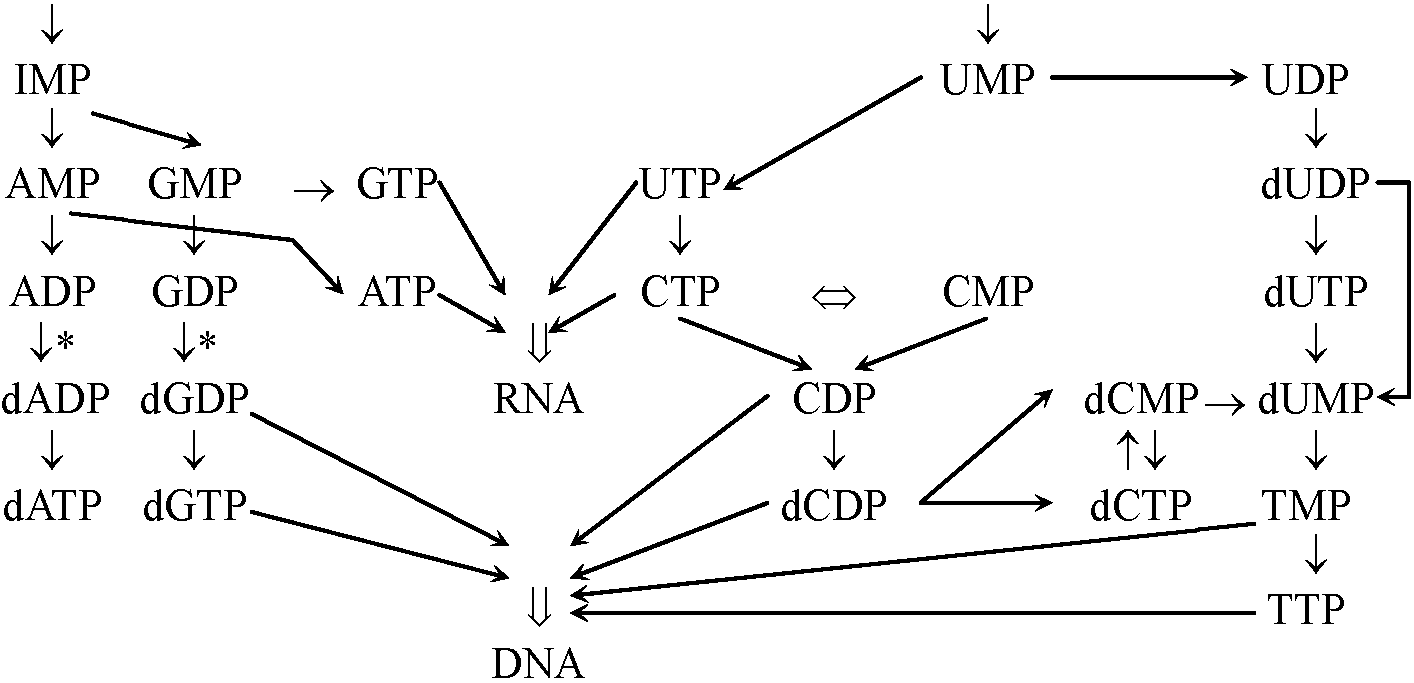
G, Aのプリンがお互いを制御する
Rat liver, hepatectomy: 肝臓一部切除し再生regeneration過程観察 Vicia favaの発芽抑制機能 → 給水するとribonucleoside-S'-diphosphate reductaseを作る |
|
1920' Levene: DNA塩基はC, T, A, Gである 1922 Griffith F: 肺炎双球菌で形質転換transformation発見
肺炎双球菌
多糖類の膜(白血球から体を保護する) 無膜型 (R型, rough colony) 病原性なし。感染しても発病しない ネズミに
R型注射 → 発病せず 死亡したネズミの体内にはS型菌が存在 → 形質転換 1944 Avery OT, マクラウンド、マッカティー
R型菌をS型菌に変化させたのは、すり潰したS型菌の何が加えられた時かを調べる
遺伝子: 安定物質で種によりほぼ一定。体細胞には生殖細胞の2倍量 1952 Hershey AD & Chase M: 放射線標識法によりDNAの働き解明 大腸に寄生するT2ファージ: CHON(SP) – タンパク質、CHONP – 核酸
→ 大腸菌内に入りT2ファージを作るのに働いたのはDNA Q. タンパク質が遺伝子、DNAが酵素の役割をしている生物での遺伝情報の伝達はどのようなものか(m-RNA相当物質は考えなくても良い)。この生物にはどのような不都合が起るかQ. DNAの2重螺旋を形成しているAとT、GとCの水素結合による結合の状態を書け。この構造を考慮し、DNAの減色効果を説明せよ DNAのつなぎかえ1976 Tonegawa
________________つなぎかえ 高等生物遺伝子組換 (組替体 recombinant)遺伝子: 交差 crossing over → 組み換え recombination____________ mRNA → 逆転写酵素 → ━━━━━━━ |
→ アルカリによるRNAの分解 → ___________ マイクロインジェクション microinjection: 本来の意味ではin vivo(in plasma membrane or cell membrane)での転写システムとして開発された 細胞融合 (cell fusion)1957 岡田善雄
エールリッヒ癌細胞にHVJウイルス(センダイウイルス)を働かせると細胞融合が起こる 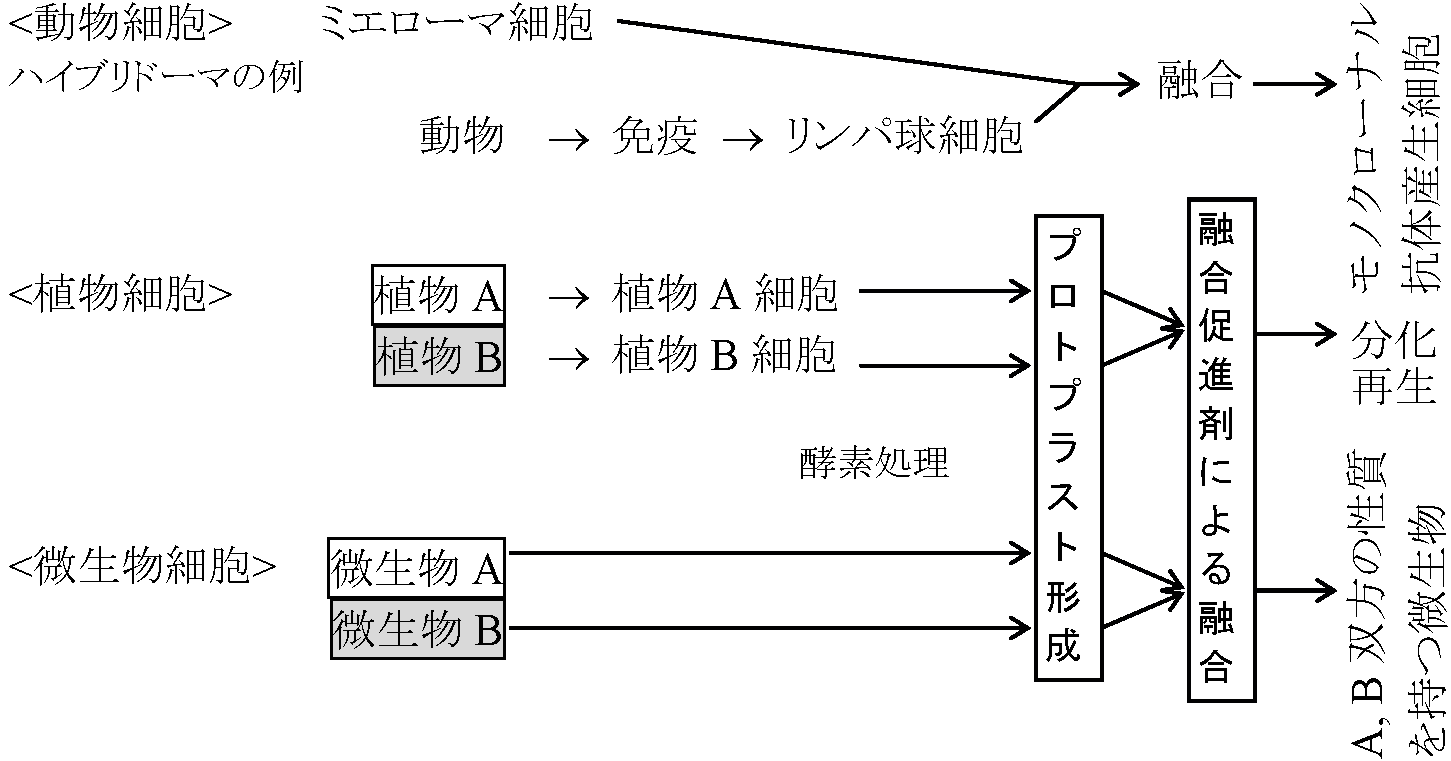 図. 細胞融合過程 |
|
1953 Watson JD & Click HC (+ Wilkins M 1962 ノーベル賞)
二重螺旋モデル DNA二重螺旋モデル double helix modelDNAの遺伝子としての性質は良く当てはまり複製の仕組み、遺伝情報の解明等、分子遺伝学発展に貢献塩基対の配列順序: 規則性はない(まったく自由)。この中に遺伝子が存在(3個の塩基配列 → 1アミノ酸) 螺旋(右巻き) 34Åで10個の塩基対が存在。直径20Å Cf. chloroplast DNA (organellaは寄生したprokaryote) → circle extrachromosomal element: 核様体でない物質 cytoplasmic element: 核様体以外の部分にある物質 Viruent virus → →
replicationがbacteriaとvirusで別_________別なbacteriaへ Lysogenic virus ↗ Virulent ↗ Virulent→ _____________溶原化 integrate ∴ coordinateで増える __________________= one replicon ex. λ, φ 分子形態1) 一本鎖DNA (single strand DNA)φX174 → circular, single strand(MW 1 × 106 dalton, 5375 bases, 9 genes判明) φX174はdouble strandのものもある(= RF-DNA)
- replicationの際形成 2) 二本鎖 double strand DNA (上記以外全て)Watson & Click model – 右巻き, Rich – 左巻きも存在a) circular double strand DNA mitochondoria DNA, chloroplast DNA SV-40 DNA – virus DNAでありながらproteinと結合(nucleosome-like strand) 1975 Helinski et al.
Plasmid (E. coli)
b) linear DNA 相補的に"糊代" 的働きをする = 粘着性末端 (cohesive end)
template phage, λ phase → 感染するとcircleになる
Ex. T4
切れて結びつく – 組換え重複する |
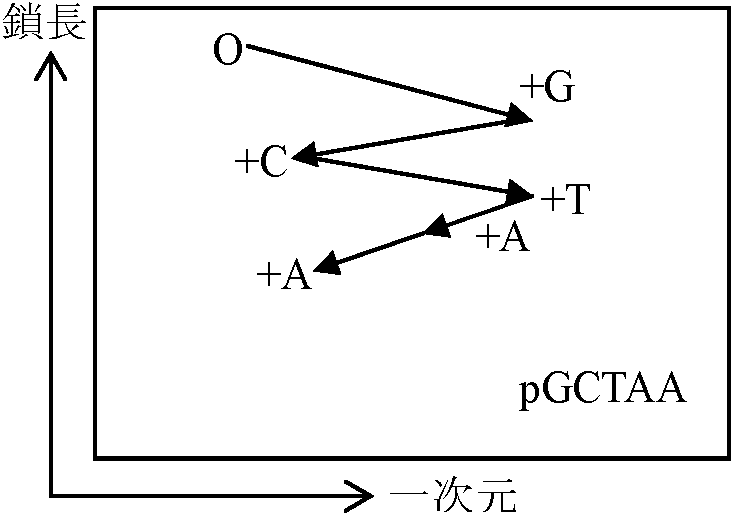
DNA一次構造決定法 (DNA primary structure - sequencing)1) Sanger method (Sanger-Lovlson)5', 3' sites付近の解析labeling
5' site: polynucleotidekinase [γ32P]ATP 2) DNA polymerase利用= classical
▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇ 3) RNAに転写
───┬┬┬┬┬─────────── DNA 4) Sanger-Loulson method (Sanger +,- method)
3' ───┬┬┬┬┬──────────────── 5'
┣━┫ ┣━┫ ┣━┫ ┣━┫ ┃
┣━┫ ┃ ┣━╋━┫ ┃ ┣━┫
┃ ┃ ┣━┫ ┃ ┃ ┣━┫ ┃
┃ ┣━┫ ┃ ┃ ┣━┫ ┃ ┃
┃ ┃ ┃ ┃ ┃ ┃ ┃ ┃ ┃
1回に100-150 bases決定可能 5) Maxam-Gilbert methodプリン → A N-3, G N-2 (メチル化)ジメチル硫酸 - 短時間処理により平均1本鎖に1箇所程度のメチル化が起るようにする → 加熱 = Gが脱メチル化 / 希酸 = Aが脱メチル化 → メチル化した部分が切れる (G > A, A > G)
→ 塩基遊離 → (ヒドラジン ピリミジン切断) C, T破壊
a) 2つの遺伝子間にごく短い空間がある場合
G遺伝子 ------->
Pro Leu Lys End
… CCA CTT AAG TGAGGTGATTT ATG TTT GGT GCT ATT …
リボゾーム結合部位 Met Phe Gly Ala Ile
<---- H遺伝子 ---->
b) 2つの遺伝子がごく僅か重なっている場合
---------- C遺伝子 ---------->
Pro Leu Ile Gly Lys Lys SerEnd
… CCA CTA ATA GGT AAG AAA TCATGAGT CAA GTT ACT …
リボゾーム結合部位 MetSer Gln Val Thr
<--------------- D遺伝子
卵アルブミン遺伝子
━▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇━ |
DNA生合成信頼度fidelity: 高いがmiscopyもある
突然変異はmiscopyによるものであり、その発生率はその種の存続に関わる問題である 原核生物Prokaryote (bacteriophyta, cyanophyta) → naked DNA 真核生物Eukaryote → DNAとそれと同量のbasic proteins (NHC, RNA) → chromatin structure
chromosomeが分かれている → linkage groupsを作る DNA合成酵素 DNA polymerase4種のデオキシリボヌクレオシド-5’3リン酸(dXTP)からピロリン酸を遊離してDNA重合反応を触媒する酵素DNA修復 (DNA repair)1) Gilbert2) 1962 Setlow & Setlow
T–T: UVがあたると*部でT=T結合 (= thymine-dimer) 色素性乾皮症 – この修復機構がないために起こる 3) 1967 GilbertDNA合成開始E. coli DNA replication1) Maaloe et al., Lark
E. coli cell cycle 80%が分裂
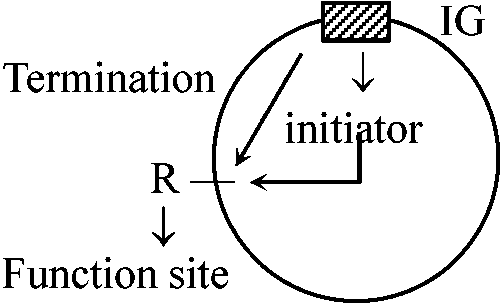
40°C → lac-, 30°C → lac+ 常温では○や○のみ複製してbのようなものが複製されない。つまり複製は別々に行なわれている 3) Replicon hypothesis: 染色体複製の調節機構に関する作業仮説1963 Jacob & Brenner
DNAで特異的なinitiatorが生成され、これがreplicatorにのみ作用して複製が開始されるという仮説 Replicon: 複製の最小単位(1つのIGを持つ) – IGを取り除くと複製できない(致死 lethal) 1963 Cairns: E.coli DNAを3H-thymidineによりradioautography
_______重金属で蒸着(リゾチームのSDSを使用) pea callus: 18 μm ↔ seedling roots: 38-42 μm (late S phase), 53 μm (early S phase) 1974 Blumental et al.
Drosophila: cultured cell = 19 m / (somatic) = 9 μm → cell cycleの時間が異なることによるのか
H2B H3 H2A H1 H4 半保存的DNA複製 (semi-conservative DNA replication)1957 Delbrueck & Stent: 半保存的複製機構の解明
Hypotheses: 1) conservative, 2) semi-conservative, 3) dispersed Vicia faba root tip 1957, 58 Meselson & Stahl: DNAの半保存的複製実験的解明
結果 DNA 14N 15N 14N15N 共にDNA取り出しsemi-conservative replication指示 1967-1970 Inman1967 Albert
|
3.5 × 104 daltonタンパク質(ssb91)発見 タンパク質の存在理由
3H(14C)thymidine → 加熱後急冷denature (H-bond切断) → 蔗糖密度勾配 → 遠心
ligase less mutant → 断片DNAのまま 1967 McKenna & Masters
PX-174-typed E.coli
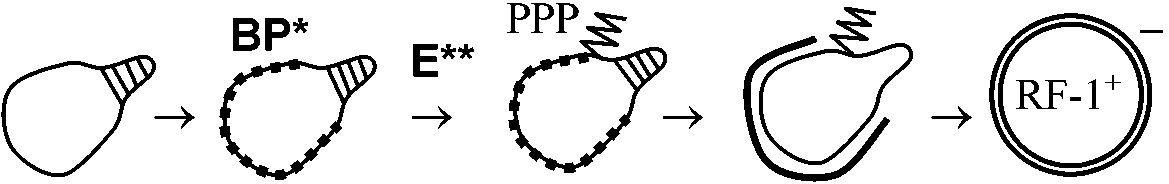
BP*: (single strand) binding protein - 切断を防ぐためDNAの周囲にタンパク質が付着
これが再び輪を作りsingle strand → 複製 (= 環状1本鎖は回転する) 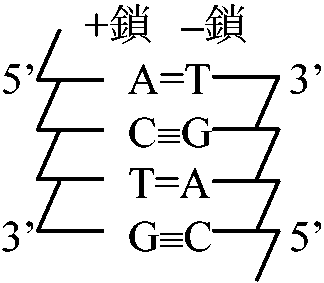 → →
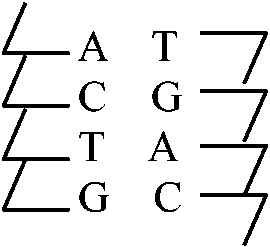 → → DNA構造_________塩基対の対合がとれ一本鎖となる  → → A, T, C, Gの4種類の塩基を持つ。デオキシヌクレオシド3リン酸がピロリン酸([P~P])を切り離して、その時のエネルギーで相補的に結合していく。DNAポリメラーゼが触媒として働く 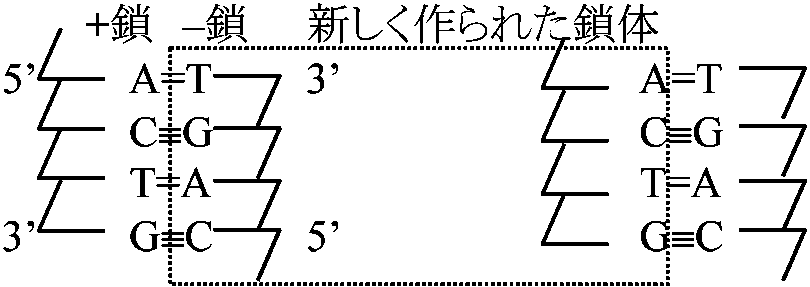 新しく作られた鎖体は古い鎖とまったく同じ塩基配列を持つ 複製 (replication)DNA複製の場所 (sites for DNA replication)仮説: Eukaryotes細胞は2n → nuclear membraneから始まる1973 Huberman, Tsai & Deich
30" labelで核中に全放射能が分布 → 合成されるものが膜についているとは限らない
isolated nuclei(核膜の外側を除去)
internal fibrogranular network: acidic proteins + RNA + lipid + (DNA) ⇒ 核のnetworkが合成の場 Def. タンデムリピート (縦列反復配列, tandem repeat): 縦列に反復コピーされた遺伝子が並ぶ配列gene ↔ 非遺伝子DNA コット解析 (cot analysis)DNA再会合に関する変数, Cot = Co (DNA濃度, concentration) × t (time)
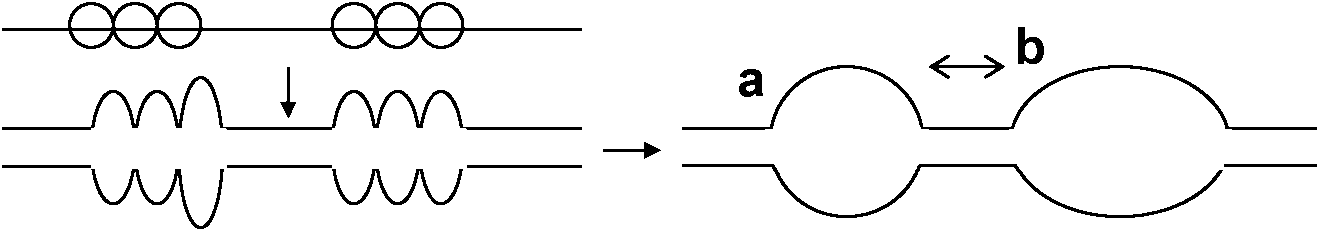
Ex. 50%再会合する場合をCot1/2と表現 → 大なら反応が遅い 細菌DNA複製 (replication of bacterial DNA)Double strand is a factor of protein, perhaps RNACircle: いくつかのループとなる → 集まって核様体nucleoid形成
1963 Cairns: DNA fiberの中に各点で複製があることを示す 1968 Huberman & Riggs
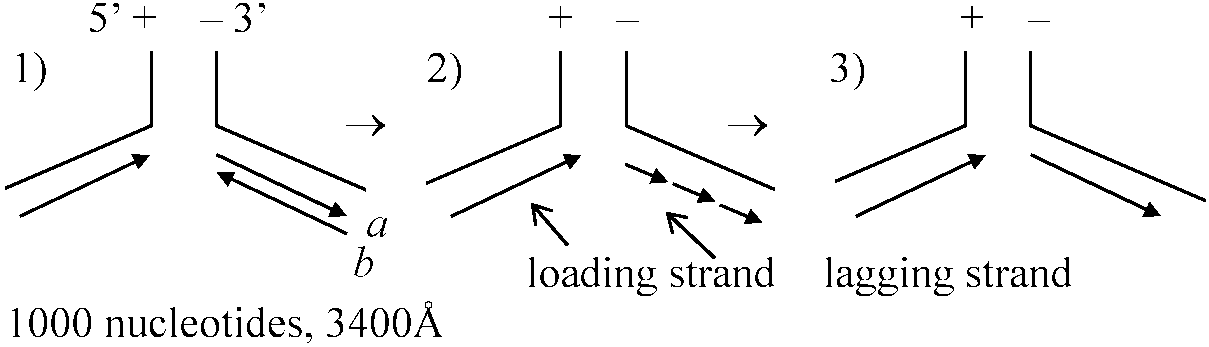
各データ比較し複製開始点をpin pointで示す バクテリオファージDNA複製一本鎖DNA (+)宿主侵入 → +鎖上に–鎖作る → +鎖と–鎖が解け新しい鎖(+に–、–に+)作られる +鎖のみが子ファージに入る(–鎖は複製時のみ活動し子ファージに入らない) |
|
1930' Beadle & Efrish Morganの所でDrosophila研究により既に一遺伝子一酵素説の考え方を持っていたが材料が複雑で証明難しく材料をアカパンカビに変える 1941 Beadle & Tatum (BeadleはMorganの弟子) 遺伝子 → 酵素 → 形質 「一つの遺伝子は特定の酵素の形成を支配し、その酵素の働きによって特定の形質が発現する」 1) 野生型アカパンカビにUV → 突然変異種(栄養要求性突然変異株)
最少培地: 野生型がやっと生活できる必要最小限の養分を加えた培地
Glu → Orn → Cit → Arg → 一処理で多数の個所が変異する確率は極めて小 1951 Beadle GW (Beadle, Tatum EL & Lederberg J. 1958 ノーベル賞)一遺伝子一初期機能仮説 「遺伝子はそれぞれ1個の初期機能を支配し、その中には酵素以外の他の生化学的な因子の形成をも含まれる」 Ex. Drosophila眼色に見られる遺伝子の酵素支配 バーミリオン シンナバー スカーレット 野生型 (朱色眼) (辰砂色眼) (緋色眼) (正常) トリプトファン → キヌレニン → 3-ヒドロオキシキヌレニン → 褐色色素 酵素V 酵素Cn 酵素St 遺伝子V 遺伝子Cn 遺伝子St バーミリオン: V遺伝子突然変異 → 酵素V作用せず色素形成がそこで停止 → 朱色眼
シンナバー: Cn遺伝子突然変異 → 酵素Cn作用せず色素形成がそこで停止 → 辰砂色眼 スカーレット: St遺伝子突然変異 → 酵素St作用せず色素の形成がそこで停止 → 緋色眼 |
Ex. カイコ眼色異常 1906 外山亀太郎: カイコの遺伝の研究 第一白卵: キヌレニンを変える酵素が変化し作用しなくなったもの 第二白卵: ヒドロオキシキヌレニンを変える酵素が作用しなくなったもの トリプトファン →a キヌレニン →b 3-ヒドロオキシキヌレニン →c 褐色色素
a: ここの酵素が作用しない異常個体は出ていない 正常: 黒眼藤色卵、異常: 白眼白色卵 人の物質代謝異常一遺伝子一酵素説で説明されるもの遺伝子突然変異 → ある酵素が作用しなくなる → 中間物質蓄積し異常生じる フェニケルトン症: フェニルアラニンをチロシンに変える酵素(フェニルアラニンヒドロキシラーゼ)の異常。フェニルアラニン蓄積し、正常経路で分解されずフェニルピルビン酸となり尿中に出る。肉体的、精神的(白痴)異常。常染色体上にあり劣性ホモで出現 アルカプトン(尿)症 alkaptonuria: アルカプトンをアセト酢酸に変える酵素の異常。アルカプトン蓄積し尿中に出る。アルカプトンはアルカリになると酸化され黒変し、黒尿症ともいう。健康に余り影響しない。常染色体上にあり劣性ホモで出現 白子(アルビノ): チロシンからメラニン色素が形成されていく過程で働くチロシナーゼが異常。弱視などを伴うことがある。常染色体上にあり劣性ホモで出現 その後 …酵素は複数ポリペプチド鎖から構成されるものがある → 遺伝子は複数遺伝子は酵素だけではなく構造タンパク質もコード ⇒ 一遺伝子-一ポリペプチド鎖(説)というべき |
|
構造: リボース + リン酸 + 塩基(ACGU)のヌクレオチド 結合様式はDNAとほぼ同じで糖–リン酸を主鎖とする鎖状構造
RNA生合成1955 Ochoa (Severo Ochoa de Albornoz, 1905-1993)
Polynucleotide phosphorylase: n·ppB (5'側にppがついている) ⇔ nPi: Mg++必要 |
1961 Hurwitz (E. coli) 1961 Weiss (E. coli) 1961 Stevens (Microsoccus lysodeiktics)
⇒ 3人ほとんど同時に発見
RNA replicase: n·pppB ⇔ (pB)n + nPPi 1966 Spiegelman, 1966 Haruna RNA virus (プラズモウイルス) → [酵素] → RNA viral RNA (active): 細胞に感染増殖 |
|
ファージmRNA: 全構造解明されている例多
MS2(f2, R17)全構造: 1RNA, 3569残基 leader-codon-tail-polyA 真核細胞mRNA
Cap構造: 5'末端にある。TailにpolyA存在 – eIF3でCap結合タンパク質がmRNA認識のために必要 安定化 mRNA (stable mRNA)1963 Tyler: ウニsea urchinのunfertilzed eggを20分間酪酸処理→ 2分 = 有核のものと無核のものができる → 両方とも発生進む ⇒ stable mRNAの存在 1964 Gross: ウニにactinomycin-D処理を行いmRNA合成を阻害
→ タンパク質合成は継続された → stable mRNA存在
8細胞期: 14C-leusine (タンパク質に入る)と3H-uridine (RNAに入る)を取りこませ追跡
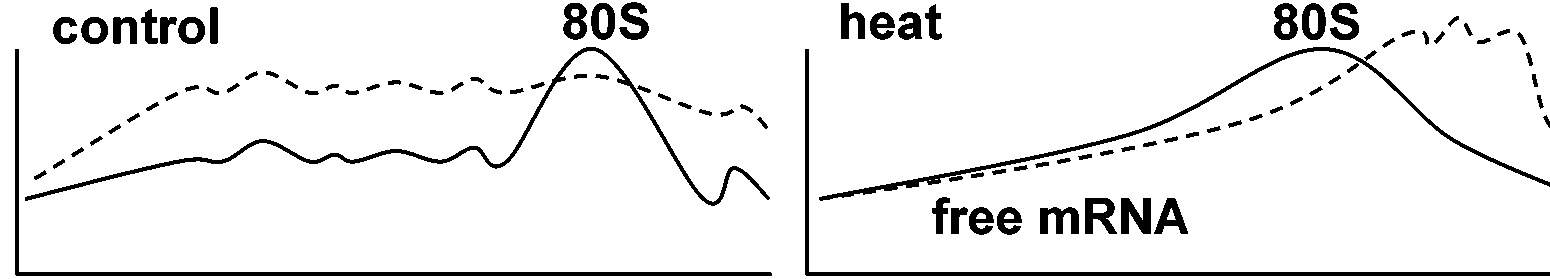
: free mRNA, protein particles = 情報粒子 informosomes
各々のRNAピークをとってショ糖遠心等を行うとタンパク質がとれる = mRNA-protein complex mRNA–polysomeの関係1. Reverse1965 Henshaw: CsClで固定したpeakパターン → complexである 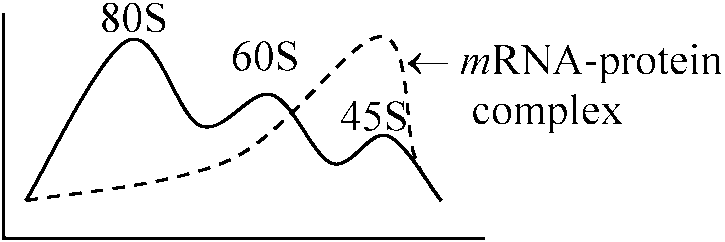 1971, 1972 Weber MJ et al.
1971, 1972 Weber MJ et al.
Animal cultured cells
Cf. mRNA labeled by actinomycin-D = 0.5 μg/ml – rRNAだけを阻害, 5-10 μg/ml – 全てのRNAを阻害 |
conserved mRNAを確認 = 乾燥種子中にmRNAがあることの実証 1968 森・伊吹・松下
ダイズを水につけ吸水させた後、ホモジェナイズして遠心後にS100を集めショ糖密度勾配にかえる RNA一次構造決定法1965 Holley: yeast tRNAAla1976 Fiers: bacteriophage MS2 - 3569 bases RNA → DNA: 逆転写酵素(DNA primary structureを決定しRNA primary structureを判断する) 1) 未標識RNAを使用する場合 (unlabelled)a) 酵素を用いて加水分解:RNase T1 = -Gp∧, RNase A = -Pyp (-Up∧ or -Cp∧) (∧: cut) b) 加水分解産物分離:2次元展開 y方向 = paper chromatography, x方向 = 電気泳動 c) Oligonucleotideの加水分解・分離i) enzymes
Pnase: -pNpN'p∧N'' → pN∧pN' d) 配列決定
oligonucleotide (DP small) 2) Sanger method (1965) |
|
タンパク質: 生体内では常に合成・分解の動的平衡状態 dynamic equilibrium
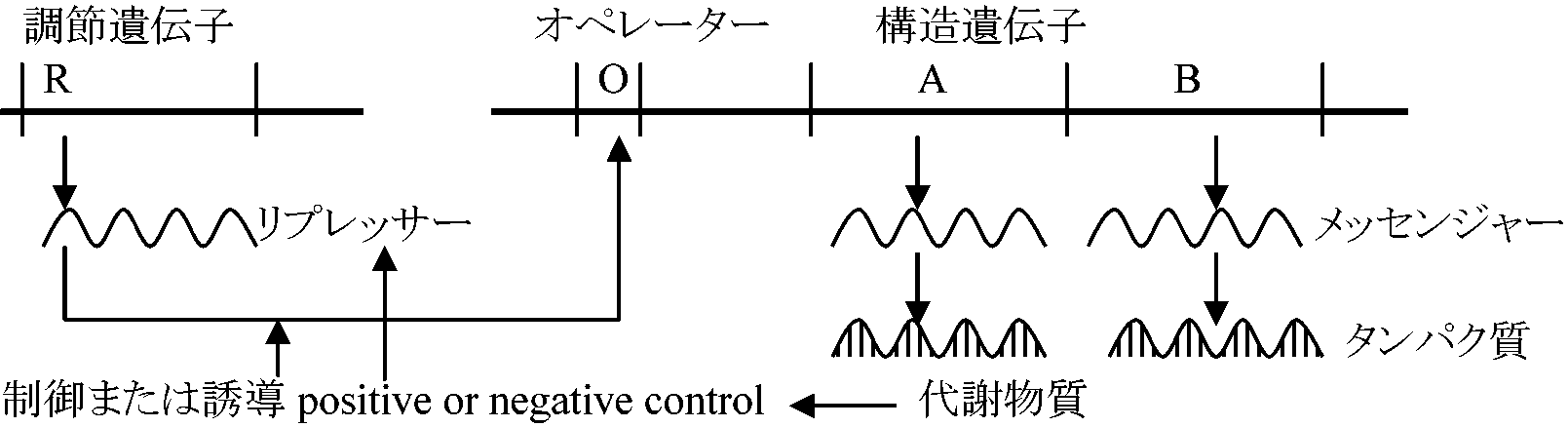
アミノ酸 → (タンパク質合成) タンパク質 → (タンパク質分解) アミノ酸 1957 Click: 一般原理 central dogma ≡ 「生命の中心説」 DNA遺伝情報 → RNA(DNA依存) → タンパク質(逆はない) オペロン (operon)酵素合成誘導: 一般に分解過程に関する酵素に見られる
E. coli: ブドウ糖をアルコール発酵により分解し生成するATPを利用する E. coli: ブドウ糖とNH3があればアミノ酸を合成するが、既にアミノ酸(トリプトファン等)を加えた倍地では合成を抑制してしまう ⇒ 既に最終産物があれば生成過程を抑制させる 1961 Jacob & Monad (1965 ノーベル賞): オペロン説 operon theory
トリプレット説 triplet1950's Coldwell & Hinshelwood仮説: タンパク質のアミノ酸配列が核酸の構造単位の配列により決まる 1952 Dounre: タンパク質合成がmRNA鋳型上で起こることを提唱1953 Watson & Click: 2重螺旋構造の発表 1954 Gamov G (Mathematician, Physician)
DNAの塩基配列とアミノ酸(aa)配列が対応すると仮定すると、塩基はAGCU(T)の4つからなるので ヌクレオシド2リン酸からリン酸を離してRNA合成酵素を発見し、それから単ポリマーの合成に成功 1957 クリック: 非重複型トリプレット説重複型は1塩基配列が決定されるとその前後のアミノ酸配列が制約を受けるため不自然
[実験] E. coliを使い無細胞タンパク質合成系作る。この合成系に擬似mRNAとしてポリウリジン(-U-U-U-U-…)を加えるとタンパク質合成のためにフェニルアラニンが選択的に取り込まれた
Mammal, sea urchin, Drosophila: 5 μm [実験] 混合ポリヌクレオチド(混合ポリマー)によるアミノ酸コードの決定
DNAにあたる …TTCTTCTTCTTCTTC…
mRNAにあたる …AAGAAGAAGAAGAAG…
[結果]
合成ペプチド鎖 …LysLysLysLysLys…
…ArgArgArgArg…
…GluGluGluGluGlu…
[考察] 3つの塩基配列が1つのアミノ酸を指定する決定的な証拠となった
1番目 2番目 3番目 mRNA遺伝暗号 genetic code (コドンcodon)縮退(縮重) degeneracy: 1アミノ酸に対し複数のコドン = 同義的synonymous暗号
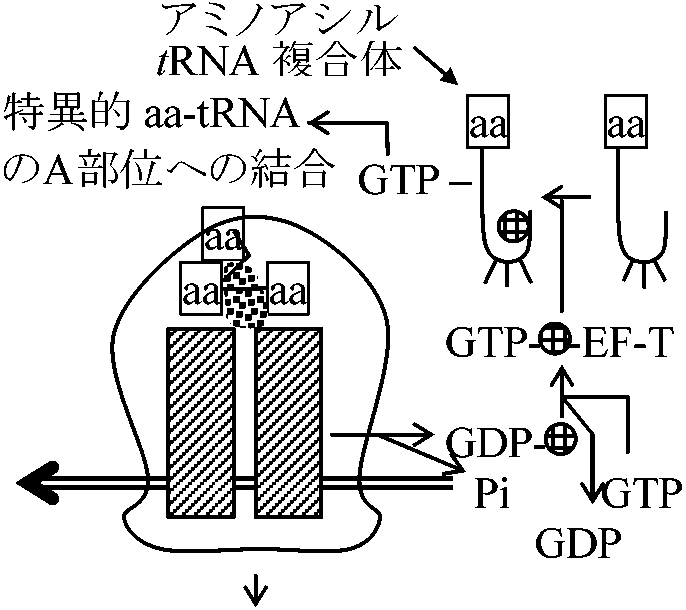
3番目の塩基は変化しても同じアミノ酸を指定することが多い ホルミルメチオニンはタンパク質合成開始暗号で合成後ポリペプチド鎖から外れることがある 終結コドン termination or stop codon: UAA, UAG, UGA読むのはタンパク質であり、これを読む特定t-RNAがある訳ではない(virus → 停止暗号2つ続くとタンパク質合成停止) ※ この遺伝暗号は地球上の全生物で同じ(ではない - マイコプラズマ)開始機構 initiation読み始め因子: IF1, IF2, GTP (IF1, IF2: 生体内mRNA → Proteinに必要)1968 野村真康: 15N-a.a.: D2O中でE. coli培養
(分子サイズ、電気的特性等の問題多) |
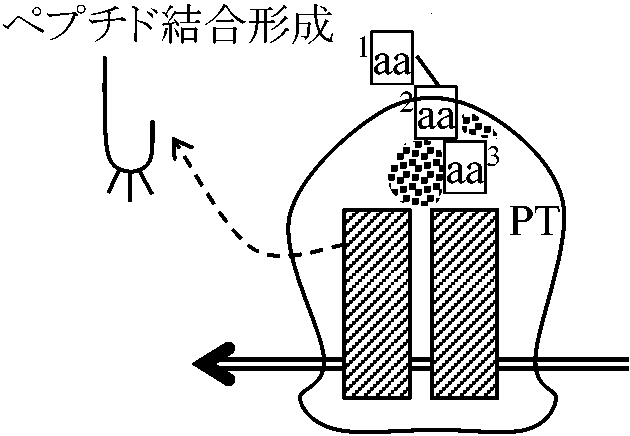
翻訳 translocationShine-Dalgarno box: AGGAGGU → 30SとmRNA結合のためAUG上流にある塩基配列、結合(挿入)のメイン部位は16S 読み終わりはDNA replicationのときと同じ
[GTP数] 原核生物(E. coli)タンパク質合成系
真核生物タンパク質合成系(E. coliとほぼ同じ) ただし、eIF種類多: cap認識等の調節が多いATPがinitiationエネルギー eIF5: 40S粒子と60S粒子を結合する酵素。リボゾーム上での因子ではない
↓ eIF2 + GTP + Met-tRNA|Met アンチセンスRNAmRNAに対し相補的な配列を持つRNAアンチセンスRNA細胞内に存在 → 相補的mRNAと結合しタンパク質への翻訳阻害 ⇒ 遺伝子発現抑制 [利用] 細胞内合成されるタンパク質量を減少させ望ましい形質への形質転換 Ex. イネ: 米粒中に含まれるアレルギー原因タンパク質の生成を抑制 |
|
遺伝子発現過程 process of gene epression
1. 遺伝子の再編成
3. RNA合成
5. タンパク質の修正 古典遺伝学 classic genetics野生型wild type → [変異剤 mutagen] → 変異型mutant type酵素変異による変異型: 不活性または生産されない なぜこの酵素がダメになったのかをbiochmical assay用い調べる = その酵素の発現機構調べる → 自ずと変異株の種類が限られてきて最終的に全ての発現機構を明らかにすることは不可能に近い改変遺伝学 reversed geneticsI. I. シグナル認識物質の同定 identification of signal DNA sequenceII. 細胞の種類Basic transcriptive function=============== DNA signal: utilized heterogenous cells Regulatory function -------------------------- cell that expressed, per se ┉┉┉━━┉┉┉=====▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇┉┉┉┉┉┉┉┉┉┉┉ ___________________________________endogenous, exogenous ┉┉┉━━┉┉┉=====▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇┉┉┉┉┉┉┉┉┉┉┉ ____________________________↓ 異なる遺伝子を入れておけばendogenousかexogenousかがわかる → しばらくすると核内のクロモゾーム中に取り込まれていくが、取りこまれる場所は定まっていない 細胞質を使った研究はここまで - 遺伝子領域まで決定できる方法ではない |
_exp. 3_exp. 1__________endo._exp.2 --███--//--░░░-███--░░░--//--███--//--███--░░░- _b______L____i_____j______m______ q____r _5 or 0__ 5___ 20___15_____100 (unit) 35___20 ███ wild type, ░░░ mutant 取りこまれた場所によってwild typeでも発現量異なる。Mutantを取りこませたとき、それが場所によって発現量が減少したのか、遺伝子を削ったために減ったのかを判断するのは困難なぜ取りこまれる場所によって発現量が異なるのか、取りこんだ遺伝子の外側領域の遺伝子を取り出してcloningし外側領域の塩基配列を決定していく。取りこみ完了細胞を1つの初期胚に取りこませ発生過程を経過させる III. 受精卵 fertilized egg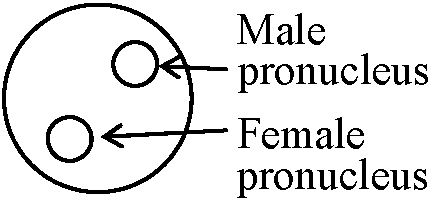 mouse/Drosophila
mouse/Drosophila
取りこまれた遺伝子は発生段階を経て脱落してしまうか、また取り残されている場合、どの細胞でどれ位の量が発現されているかを調べる → test of regulated expression → identification of signal DNA sequences Findings of the component responsible for the regulated functionある遺伝子マーカーに異常のある生物を異常のないものに入れ換えることによりその生物を救える
= gene rescue ゲノム編集特定の遺伝子配列を特異的に認識するシステムTALE-NucleaseCRISPR-Cas9guide RNA (rRNA, tracrRNA): 相補的配列を含み標的配列に結合するRNACas9: guide RNA に結合し標的配列を切断するタンパク質 |
|
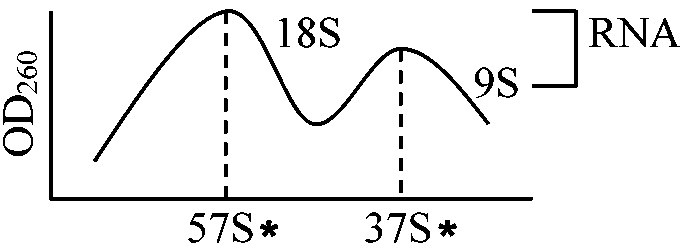
1968 Brown (1969 鈴木): Xenopus laevis, ribosomal RNA genes
specific gene amplification in oocyte → DNA-RNA hybridization
(Suzuki & Brown 1972) Isolation of sil fibroin mRNAFinger printUse DEAE-Sephadex
Detection of fibroin genes1887 Boveri: Parascaris equorum
体細胞染色体は成長段階で不必要な部分が切り捨てられる 発見: 体細胞系列で染色体が減る 1972 Suzuki, Gage & Brown
2 cells period 4 cells period サザンブロット (Southern blotting)1975 Southern EM: サザンブロット開発
ゲノム中の特定配列の位置・コピー数・構造変化を物理的サイズとし検出
2種類の2本鎖混ぜる - 似ていればハイブリダイゼーション起こる (Suzuki & Ohsima 1977, Ohsima & Suzuki 1977) Isolation and characterization of fibroin gene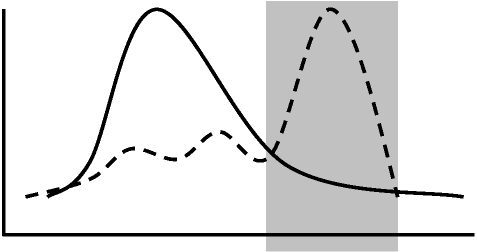 firroin gene - 0.004% of the genome
firroin gene - 0.004% of the genomeBombyx mori 2.5 × 10-5 = 0.52 pg (cloning容易) General ~ × 10-6 = 3 pg (ex. mouse) Act-D/CsCl (Act-D: Gに付着し比重が軽くなる): geneを短い断片にしてこれをcolumnにかけるGを多量に含むgene → firboin gene これを抽出すれば濃縮可能 1000倍濃縮 純度0.4%
→ cloning ⇒ 転写開始末端必要 = より長い遺伝子鎖gene chain欲しい 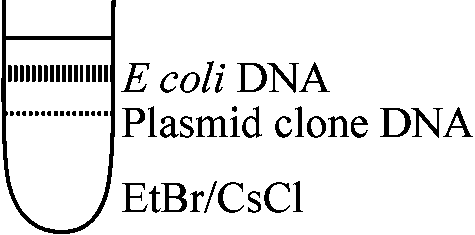 Cloning法: E. coli中にplasmid (vector)移入し、それをCM中へcultureするとvector geneだけが最大1000倍程度に増殖
Cloning法: E. coli中にplasmid (vector)移入し、それをCM中へcultureするとvector geneだけが最大1000倍程度に増殖
遺伝子構造解析 (structural analysis of the genes)1981 Busslinger, Moschonas & Flanell
21 bpのところにmutation[normal = G, β+ = A]
ex. 1 int. 1 ex. 2
Intron 5'末端や3'末端の遺伝子が異種でも混在conserveされる。GがAに換わるとintron 3' siteとの類似性高くなる。本来intron 3' siteはexon 1の3'末端を見分ければよいが誤って21 bp付近を認識する可能性高
Transcription initiation 実際の転写開始の信号 (Ex. AGT) = promoter Hybrids arrest Ex. 数の少ないmRNAのクローン 抗体でpolysomeを落としmRNAをとる。しかし、この時期のmRNAも同時に取れるがそのままcopy DNAを作らせる → 調べるとintronのないものもある (ex. histone H1, H2A, H2B, H3,H4などには無い) 生体外翻訳系利用 (use of in vitro transcription system)Type I gene =ETS (external transcribed sequence) ITS (internal transcribed sequence) Coding region Unique geneでなく、この3つを単位にNTS (non-transcribed spacer)となり転写されない。r-RNA large unitになる25-28Sの方はintronがあるがsmall unit r-RNAの方にはない。Drosophila melanogasterでは2500, 1200のcoding region間に50% 500, 1000, 5000のintron、15% 3000, 4000のintron、35%の非イントロンがある 反復配列repeated sequence: 長さ不均一 → NTS長変化 Type II gene: modified RNA polymerase II genesPosterior silk gland extract (Tsuda & Suzuki 1981)
比べるもの同士を1本の試験管に入れると +514: A >> B (dominance assay or competition assay) = enhancer-like element ⇒ enhancer element (DNA) → SV40, plyoma etc.に似た性質がある
P.S.G. cell
Fibroin gene
Sericin gene
Ad geneはHela extractで良く発現 ↔ PSGではそれ程でもない Hela
PSG
hnRNA → (processing) mRNA = 初期転写産物 primary transcripts |
一般的特性
0 exon-intron
2 non-translated region (GT-AG mule)
リプレッサー repressor: DNAのプロモーター近傍に位置しオペレーターに結合し遺伝子発言を抑えるるタンパク質性転写調節因子 1) Exon-intron junction: IntronのGT-AG mulePhaceoline (タンパク質)
Exon
Intron
Exon 2) Pormoter原核生物ではprobnow box (prokaryotes promoter)が知られる“TATPuATP”配列。真核生物ではこれに対応するものがGoldbery-Hogness (TATA) boxと呼ばれる。これらの信号のあと原核生物では5-6、真核生物では29-35塩基後に転写開始点initiation pointがある。KB cell (Reader 1979)とHela cell (Marley 1980)でin vitro転写(DNA → RNA)可能(Reader 1979) – 転写開始点が明確になったことによる
de in vitro: analysis of product … TATA … → … TAGA … 機能するが効率落ちる Hypersensitive region: Rabbit β-globlin gene-40から-100 ⇒ TATA boxより優先される決定因子 Modulator1980 Brinstel: Sea urchin histone gene H2A
-184 ~ -524: E-region, +1: efficient-beginning position Enhancer (Activator)SV40 – circular → straight____72b____72b______21b 21b 21b_____TATA box___mRNA ----░░░░░░░░░░░------██--██--██---------█████------████------ __repeated sequence = enhancer (GC rich)
1. short base sequence Termination signalAATAAA (poly A): 1000倍 (cf. re =reverse, tr = transcriptase, o = oncogene)Hetro virus (RNA virus) 60-70S single strand RNA
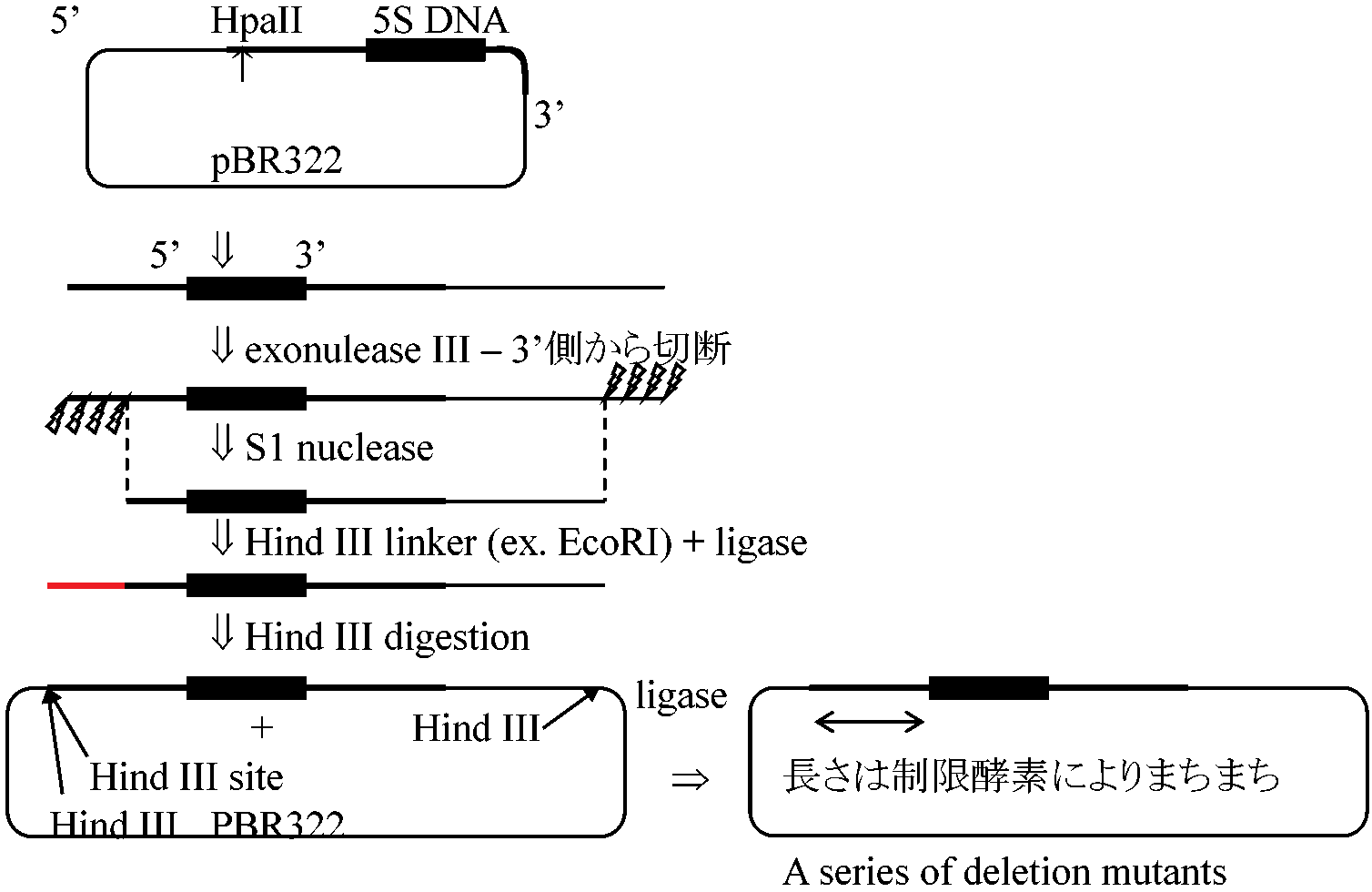
______R U5____________U3 R___3 (3 primer), 5 (5 primer) splicingに関するsmall nucleous RNAでキャップ構造を持っていないもの  1980 Brown D & Sakoyn: Xenopus 5S RNA
1980 Brown D & Sakoyn: Xenopus 5S RNA
Transcription of 5S DNA: ~20000 copies/genome
"in vitro genetics" proposed by Brown
次に3’側からcut (Bugenhagen, Sakonju & Brown 1982)
+51から+90の間に何らかの転写開始要因がある: 遺伝子制御 = control region ≈ promoter
======================
-------████------- + nuclear extract (5S factor + histones)
left____GGTCTAGTGG
tRNALys, histon, polymerase III → chromatin reconstruction |
☛ 細胞質遺伝
|
Def. 細胞小器官の遺伝子(DNA) - 非メンデル遺伝: 核遺伝子と独立に複製
受精卵細胞質 - 雌性配偶子由来 → 遺伝形質は常に雌親と同 ☛ ミトコンドリア ミトコンドリア遺伝子 (mitochondrial gene, mt gene)Eukaryotes: Cu, Zn ⇔ mt: Mn = Prokaryotes: Mnmtが有するDNA情報量 < 核DNAの1/10 1. シトクロム酸化酵素 (cytochrome oxidase)サブユニット(分子量): 同調的に合成される = I(40000) II(22500) III(15000) IV(11200) V(9800) VI(7300) VII(?)
II = mtDNAから
N末端には疎水性(脂溶性)アミノ酸が多い L8S8 × 16サブユニット → L8 = mt DNA, S8 = 核DNA 3. RNA: rRNA (ribosomal RNA), tRNA (transfer RNA)葉緑体遺伝子 (chl gene, chloroplast gene)プラスミド (plasmid)Def. 核外存在 - 細胞分裂で娘細胞へ引継ぐDNA分子a) 自己複製単位: 自身複製replication、segregation指令 染色体DNAから独立し複製 (染色体と同調し複製) b) プラスミド遺伝子 plasmid gene: 細胞自身の生殖(増殖)にはなくてもよい細菌生存に関係なくある程度の数だけ増殖 → 協調関係 Def. エピゾーム episome: 主染色体外遺伝子 extra-chromosome で寄主に潜り込めるもの (Jacob & Wollman 1961)Ex. sex factors, colicin, λ-phage (≠ ミトコンドリアDNA、プラスチドDNA) c) プラスミド消失: 時々消える人為的例: acridin (Hirota 1960), SDS (Tomoeda 1968), Ni, etc. d) プラスミドDNA: 環状二重螺旋 circular double helix= 閉じたループ closed loop |
構造: 共有結合部分がある – スーパーコイル super-coil or helix 構造
Ex. 一ヶ所切断すると、プラスミドがocDNA (open circular DNA)となるためcccDNA (covalently closed circular DNA)であることが解析できる
______DNase 1.2-1.5 MDt (106 dt)が最初のプラスミド。例外的にpBR345 (0.7 MDt)という極小プラスミドが知られるが、これはdimerをなし結果的には1.4 MDtである。最小プラスミドは1.2-1.5 MDt位といえる F因子(F plasmid, fertility factor): 性決定 - 50タンパク質
F+ strain: 独立した形で保有しているもの
主染色体の耐性能力としてはリボゾームタンパク質構造の変形による程度の能力しか有していない。ところが、R因子は抗生物質に変化を起こさせる(Ex. アセチル化による抗生物質不活性化)ことで耐性獲得。しかもR因子のR+からR-への伝播は早い
このプラスミドは他細胞上に伝播可能なものと不可能なものがある
伝播可能 Col B, Col E F pili形成に関与 分解プラスミド degradation plasmidsミルク中: Streptococcus lactis, S. cremorisfree amino acids – few カゼインcasein (protease), ラクトースlactose (lactase)等の形で存在 – 遺伝子はプラスミド上 E. coli, Salmonella: ショ糖発酵 – 遺伝子はプラスミド上 Pseudomonas: 多種多様な物質を餌にする naphthalene – NAH plasmids, salicylate – SAL plasmids, Camphor – CAM plasmids, n-octane – OCT plasmids, toluene, xylene – TOL plasmids のように資源化能を有するプラスミド |
Fertilized egg利用assay systemMouse metallo thionein I gene: 金属が細胞内にたまると活性化され金属を排除(解毒)する役割を持つ遺伝子のpromoter, regulator reigons – thymidine kinase gene
Mouse PMK +K enzyme + K mRNA (■: 発現した) (Wanger et al. 1981, Constantini & Cacy 1981,
human growth hormone本体部分detect → mouse
#pMGH/ mRNA/ Growth Growth Copyする数多いほど発現量多 - Growth hormoneは一般に脳で合成される。しかし、実験では働くpMTのpromoter + 上流部を持っているから生成されるに他ならない Spradling & Rubin (1982), Rubin & Sprading (1982)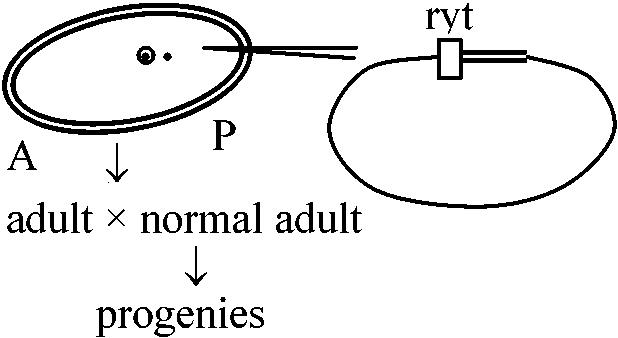
ショウジョウバエ卵にgene注入 |
DNAマーカー DNA markerできれば…少労力で多遺伝子座(locus)を一度に調査できるマーカー選択+ 対象分離集団選択や遺伝実験系統の活用がマッピングの効率化の鍵 実験手法に基づく分類1. サザンハイブリダイゼーションRFLP (restriction-fragment-length polymorphism, 制限酵素断片長多型)制限酵素で切断されたDNA 断片長が種内個体間で異なる(= 多型)こと DNAフィンガープリント: DNA配列 = 指紋の様に個体特有
Ex. 犯罪捜査で個人特定。遺伝病診断
タンパク質特定部位切断制限酵素 - 断片はタンパク質毎に独特の特徴 2. PCR (polymerase chain reaction, PCR)Table. Characteristics of molecular marker (Rafalsky & Tingey 1993)
|
画期的DNA増幅法 (Kary et al. 1980 = 1993 ノーベル化学賞)
|
1986 Mullis et al.: PCR法開発 1989 Science "The Molecule of the Year" (特許権$3億) 微量遺伝子をin vitroで純粋に十分量まで増やす → 様々な解析 原理: DNA = 相補的2本鎖 → 変性させ1本にしたDNA
+ dATP, dCTP, dGTP, dTTP + primer + DNA polymerase
DNAを数時間で100万倍に増幅 アンプリコン(増幅産物) amplicon: PCRで増幅されたDNAのこと 伸長反応: プライマーのアニーリング後、DNA polymeraseによりDNAの相補鎖を合成する反応過去の難点: 94°C = DNA変性温度 ↔ DNA polymerase失活 ⇒ 1サイクル毎に酵素を足す必要
温泉中の高温耐性菌 → 耐熱性DNAポリメラーゼ(Taq DNA polymerase) = 94°Cでも失活しない
3段階温度変化を周期的に行う(サーマルサイクラーで完全自動化)だけ プライマー: 鋳型DNAに相補的な塩基配列を持つ合成オリゴヌクレオチド(短い1本鎖DNA, 10個程度)。増幅領域挟むよう2か所設計 = ペアをforward/reverseまたはsense/antisenseと呼ぶ ゲノムDNA塩基配列上に個体間差存在 → 増幅DNA断片の種類・大きさに違い(= RAPD) プライマーダイマー: プライマーを鋳型とし合成される目的外の増幅産物。プライマー同士で相補配列存在 → その部分がアニーリングし通常のPCRと同様に指数関数的に増幅 → 増幅サイズ短いため目的増幅産物よりも増幅効率高いことがある → 検出感度低下 インターナルコントロール, IC: PCR阻害の有無判別が目的試薬にIC添加された系: IC増幅 = PCR阻害はないと判断 ⇔ ターゲット、ICともに増幅認められない = 偽陰性の疑い 鋳型(テンプレート) templatePCR: PCR増幅の元となるDNART-PCR: 逆転写反応(RT, reverse transcription)に続いてPCRを行う方法 RNAはDNA polymeraseの鋳型とはならない → RNAをDNAに変換 転写: DNA → RNA ⇔ 逆転写: RNA → DNA cDNA (complementary): 逆転写でRNAから変換されたDNA = PCRの鋳型 ⇒ RT-PCR: RNAをPCRで増幅する手法 マスターミックス: まとめて調製した反応液反応液調製時に、1反応分ずつ混合するのは操作性が悪く、正確性にも欠ける ↔ 鋳型以外の反応液(マスターミックス)をまとめて調製 リアルタイムPCR (real time PCR, quantitative PCR, qPCR)PCR反応液の中に予め蛍光標識プローブか蛍光色素を添加し、リアルタイムで目的遺伝子の増幅をモニタリングする利点: 電気泳動不要 = 迅速・簡便・定量的解析可能 増幅曲線 amplification plot: PCR増幅過程を蛍光物質用いモニタリングサイクル数(X軸)-蛍光強度(Y軸)関係をみる曲線 Ct値 (Cq値, threshold cycle): 増幅曲線-閾値交差するサイクル数
= PCR増幅産物がある一定量(閾値 threshold)に達した時のサイクル数 クエンチャー(消光物質): 蛍光物質の蛍光を抑制する物質 |
定量PCR: PCRにより定量的に検出すること(普通はqPCR装置用い行う)
サイクリングプローブ (qPCR蛍光検出法)サイクリングプローブ: RNAとDNAからなるキメラオリゴヌクレオチド一末端が蛍光物質で、他末端がクエンチャー物質で修飾 インタクト = 蛍光発しない → PCR増幅産物とハイブリッド形成 = 反応液中に含まれるRNase HによりRNA部分が切断され蛍光発するサイクリングプローブのRNA付近にミスマッチ存在 → RNase Hによる切断なし → 配列特異性高い検出が可能 - SNPsタイピング等に最適 インターカレーター法 (qPCR蛍光検出法)2本鎖DNAに結合し蛍光発するインターカレーターを反応液に加え、増幅に伴う蛍光を検出する方法。PCR産物増加に伴い蛍光強度は上昇Tm値 (融解温度, melting temperature): 2本鎖DNAの半分が変性し1本鎖DNAとなる温度 プライマーのTm値はPCRのアニーリング温度を決定する参考 融解曲線分析 (melting curve, dissociation curve): インターカレーター法によるqPCRで、PCR増幅産物のTm値を確認する方法PCR反応後、反応液温度を60°Cから95°Cまで徐々に上昇させ蛍光値をモニタリング → PCR産物が2本鎖を形成した状態では強い蛍光を検出 → ある一定温度(Tm値)に達すると1本鎖に解離し蛍光値が急激に低下 → Tm値はPCR産物の長さやGC含量により異なるので、目的増幅産物とプライマーダイマー等の増幅産物を区別できる EMA-PCR: PCRにより生菌由来DNAを選択的に検出する方法EMA (ethidium monoazide): DNA結合インターカレーターの一種
→ 可視光照射によりDNAに共有結合 電気泳動でDNA検出時に使用。発癌性(取扱注意) UNG (uracil-N-glycosylase): PCR増幅産物のキャリーオーバーによる偽陽性を抑制する酵素dTTPの代わりにdUTPを含む基質を用いPCRを行い、増幅産物にU塩基を取り込ませる。このPCR産物に対しUNG処理を行うと、U塩基の部分で加水分解され、キャリーオーバーを防止 マルチプレックスPCR複数のターゲット領域を同一の反応液中で同時に増幅すること
増幅産物の電気泳動: 各ターゲットを増幅サイズにより区別し検出 コンタミネーション contamination= PCRの鋳型となり得るDNAの混入高感度検出法 → 微量DNA混入 → 増幅され判定結果に大きな影響 エアロゾル: DNA溶液のエアロゾル発生 → コンタミネーションの原因高濃度DNA溶液の取扱には注意 エリア分け: 実験エリアを区分 = コンタミ対策PCR反応液調製、検体からの鋳型DNA調製、PCR反応液への鋳型添加、電気泳動の場所を別にし、機器・器具等も部屋毎に用意 → 実験設備内で試料の流れを一方向にし反応液への増幅産物混入防ぐ 遺伝子検査ある生物種固有塩基配列(遺伝子)を検出する検査 - PCR法良く使用培養法に比べ、迅速に結果が得られ検出感度高い MIG-seq法= multiplexed ISSR genotyping by sequencingPCRのみでゲノム縮約ライブラリー構築 ISSR = inter-simple sequence repeat 長所: 低品質/微量DNA利用可能、迅速(3日)、容易(PCR2回 + NGSラン)安価(750-1500円/サンプル) 短所: 数万座以上の解析に不向き、SSR近傍の配列解析に限定 応用: クローン識別、遺伝的構造・多様性、種間関係 |
|
熱抽出法: 熱処理によりDNA抽出する簡易的方法。主にグラム陰性菌(真菌やグラム陽性菌では、十分量のDNAが得られないこと多く適さない) アルカリ熱抽出法: 簡易的なDNA抽出法。アルカリ条件下で熱処理を行い、中和後、遠心上清を回収 目的: 生体中からの核酸抽出原理を理解 [本来抽出核酸の性質を調べるが、既製核酸を用い核酸の定量及び定性(特にGC含量)測定を行う] 1. Schmidt, Thannhauser, Schneider法による核酸抽出Step 1. 酸可溶化物の除去(よく冷やしながら行う)
0.5-5 g(w/w)のsampleを冷えた乳鉢mortar中でpestle乳棒を用いて、すり潰しながら10-20 mlの冷やした5% PCAを加え良くかき混ぜる a-b → 3回繰り返す
c) pptを得る
a) Step 1のpptに95% Et-OHを10-20 ml加えて良く混ぜる → d-e: 3回繰り返す f) (真空ポンプかドライヤーで) pptの溶媒をとばしsampleを乾燥させる (ドライヤーではsampleが吹き飛ばないよう注意) Step 3. RNA抽出a) Step 2の乾燥pptを0.5 N KOH 37°C, 16-20 hrで処理 (加えるアルカリ量はfresh sampleあたり10 ml程度) b) 冷やしながら6 N HClで中和(濃PCAによる中和も可能) DNA proteinが肉眼で見える
c) 10000 rpm for 5 min
Step 3のpptを5 mlの5% PCAに懸濁させ90°C 15 min放置(塩基遊離) 長時間置くと塩基外れるので注意。20°C, 37°Cの2種の温度で培養したものを別々に抽出し比較 結果: 抽出できたが乾燥重量測定に失敗した 2. Ceriottiの方法によるDNA定量方法(標準曲線作成)DNA、0.04%インドール酢酸(難溶時は温水使用、冷所保存)、濃塩酸(SG 1.19)、クロロフォルム(高純度) 材料: 子牛胸腺DNA |
手順
抽出DNA sampleは2-3種類の濃度について同様の操作を行い定量。この方法では3.3%以下のPCA混入は影響が無い。又DNA濃度が低くPCA混入が大きくなる場合には濃縮する 3. DNA塩基組成分析
補: 1. 6 mg/0.1 ml 2. spotは正確に置き、乾燥後、展開を15-20 cmまで行う 3. 250-260 nmの単一波長が良い。今回NISO4·7H2O 3.5 g, CoSO4·7H2O 10 mlにしたものがca 260 mnを選択的に通したためそれを利用した
展開液及びRf値 A G C T
検出には少なくともRf値が0.1以上の離れた展開液を用いる必要がある
→ 7.5 (μg/ml), 15, 21, 24, 27 → 吸光度 = 0.088, 0.167, 0.213, 0.247, 0.250
稀釈倍率 1st 2nd Av μg/2 ml sample 得たDATAから2972.5 μg/2 ml sampleが得られる 4. イオン交換カラムクロマトグラフィーによるDNA塩基解析材料: CoH thymus DNA樹脂: Dowex 50W-X4, 200-400 mesh H+ for m 手順: DNA 2-3 mg (3 mg)を6N HCl 2 mlに溶かす → 100°C 3 hr加水分解 → 冷却後、水加え6 ml → DNA 1 mg相当をカラムに載せる → 2 N HCl溶出(流速: 30 ml/hr = 4 ml/tube) → 各塩基分画集め体積とA260計りモル数算出 準備: 2N HCl、Dowexを詰める(空流を行い流速計っておく) |

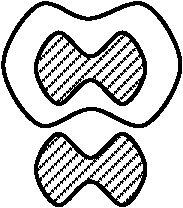 有膜型(S型, smooth colony)
有膜型(S型, smooth colony)


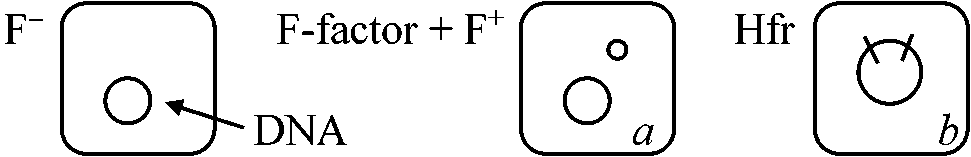

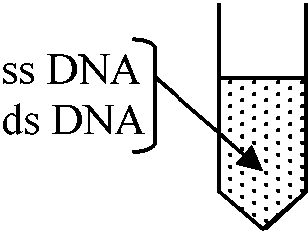 ⇒ cellulose: タンパク質付着 - それを調べる
⇒ cellulose: タンパク質付着 - それを調べる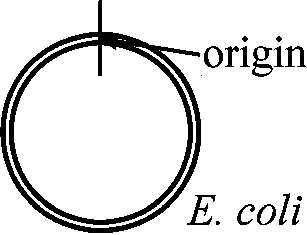 74上にilr gene (A B C)確認: ここから遺伝子複製開始
74上にilr gene (A B C)確認: ここから遺伝子複製開始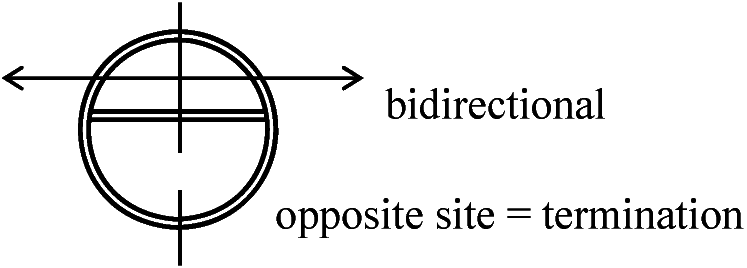
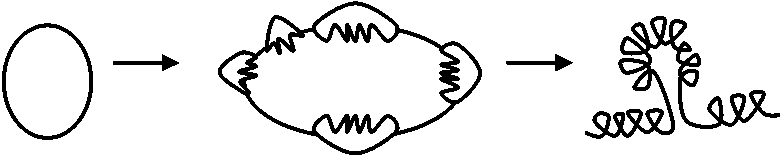
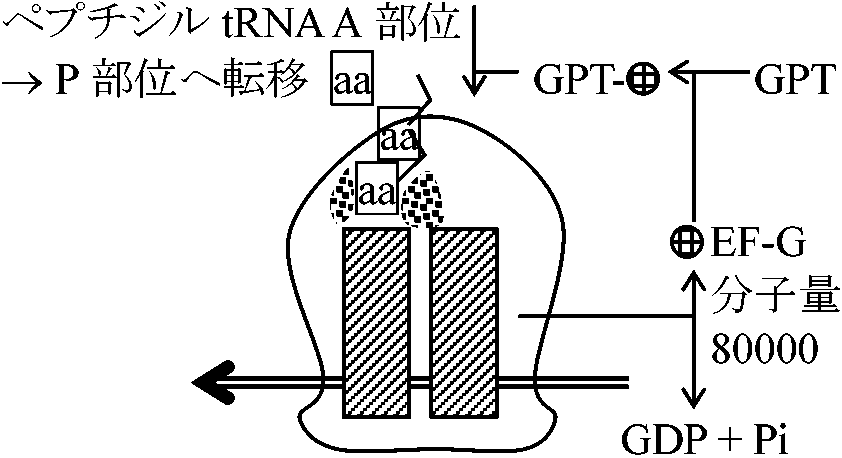
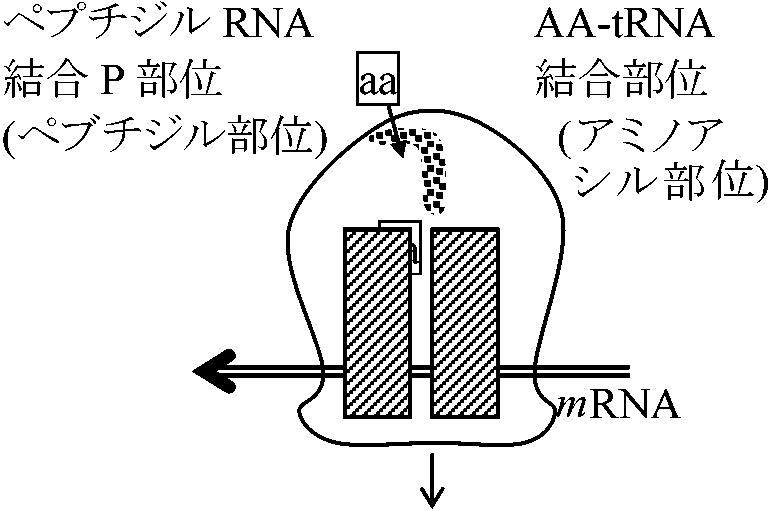 成長しつつあるポリペプチド鎖: 末端tRNAでタンパク質合成部位につないでいる。(1)成長しつつあるカルボキシル末端には常にtRNA分子がついている(N末端→P末端に合成)。この末端のtRNAがP部位またはA部位に結合することが、成長しつつあるポリペプチド鎖をリボゾーム上に保持する主な力となっている。
成長しつつあるポリペプチド鎖: 末端tRNAでタンパク質合成部位につないでいる。(1)成長しつつあるカルボキシル末端には常にtRNA分子がついている(N末端→P末端に合成)。この末端のtRNAがP部位またはA部位に結合することが、成長しつつあるポリペプチド鎖をリボゾーム上に保持する主な力となっている。
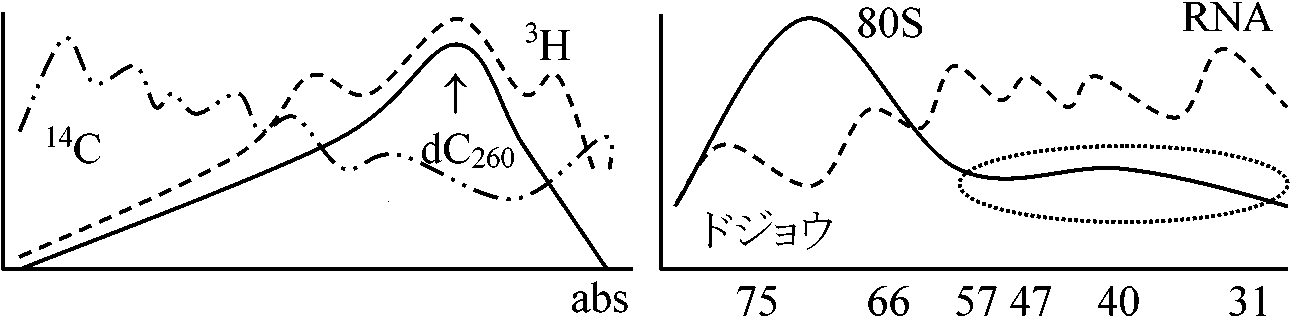
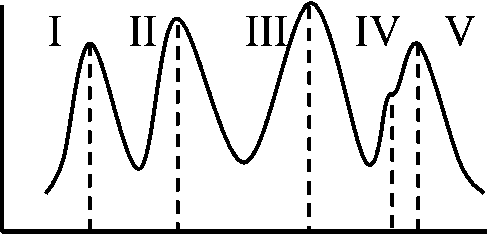 4つのピーク: 理論的にはIVのピークが消えたグラフとなる
4つのピーク: 理論的にはIVのピークが消えたグラフとなる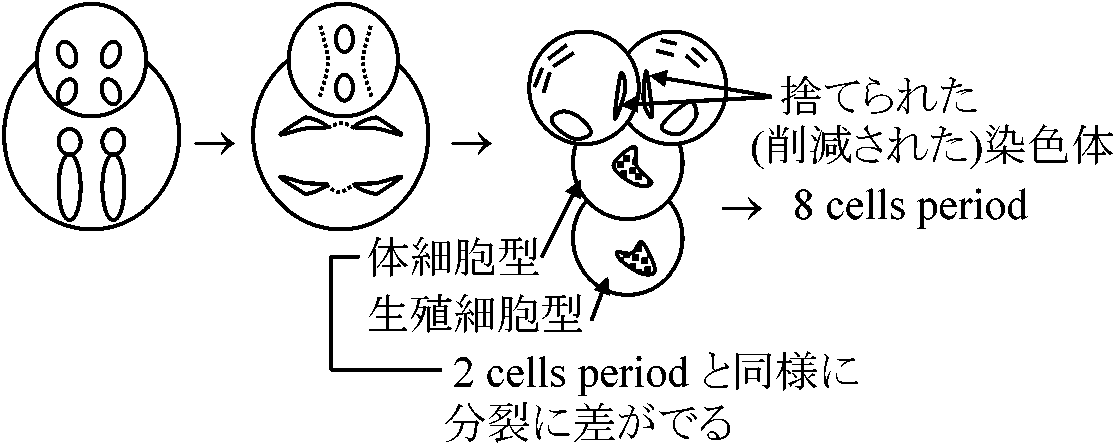
 ⇒ 7/10000の頻度でfibroin geneがとれる
⇒ 7/10000の頻度でfibroin geneがとれる